Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
DSB Double-Strand Breaks causate da radiazioni stress ossidativo
farmaci

2
DSB e CROMATINA Higher-order chromatin packaging is a barrier to the detection and repair of DNA damage DSBs induce a local decrease in the density of the chromatin fibre, in addition to altering the position of nucleosomes DSBs also elicit post-translational modifications on the protruding histone tails

3
Chromating remodelling and DSBs
RSC remodels the DSB chromatin The PIKKs Mec1 and Tel1 phosphorylate H2A(X), and RSC accumulates in the regions flanking the DSB SWI/SNF is recruited and remodels the donor template chromatin
, and RSC. accumulates in the regions flanking the DSB. SWI/SNF is recruited and remodels the donor template chromatin.")
4
RSC complex RSC (remodels the structure of chromatin)
ATP-dependent chromatin-remodelling RSC can mediate nucleosome sliding, alter histoneDNA contacts and remove histones from DNA. The chromatin-remodelling activity of RSC is important for transcriptional regulation of genes that are involved in stress responses and cell-cycle progression

5
Chromating remodelling and DSBs
Phosphorylated H2A recruits cohesin, which helps to bridge interactions between sister chromatids The HDAC complex Sin3–Rpd3 removes acetylation from H4. The protein kinase CK2 is also recruited and this phosphorylates H4 The INO80 complex enters the region of the DSB and removes some nucleosomes.

6
MODIFICAZIONE ISTONI Eukaryotes have several histone variants, which, as a result of their altered amino-acid composition, can affect both the structure of individual nucleosomes and the ability of nucleosomes to form higher order chromatin structure The earliest and most robust modification induced by DSB is phosphorylation of the histone H2A variant H2AX on its extended C-terminal tail. Within seconds, phosphorylated H2AX (known as γ-H2AX) spreads over a region spanning thousands to millions of bases surrounding a DSB
 spreads over a region spanning thousands to millions of bases surrounding a DSB.")
7
H2AX extended Ctail

8
PIKKs = phosphatidylinositol-3OH-kinase -like kinases DNA-damage sensor proteins Ku70–Ku80, MRN, RPA TOPBP1

9
DDR signal spreading DDR proteins initially accumulate at DSB sites and then spread at distance via a positive feedback loop involving MDC1, which binds gH2AX, the MRN complex, and ATM kinase, which phosphorylates additional H2AX molecules further away from the break site. Spatial organization of DDR protein accumulation at DNA DSBs. (A) (B) Regional distribution of DDR proteins around DSBs. Factors involved in ATR signaling accumulate proximal to the break site on ssDNA generated by DNA end resection, while ATM signaling factors localize on flanking chromatin regions
 (B) Regional distribution of DDR proteins around DSBs. Factors involved in ATR signaling accumulate proximal to the break site on ssDNA generated by DNA end resection, while ATM signaling factors localize on flanking chromatin regions.")
10
Spatial organization of DDR protein accumulation at DNA DSBs
Spatial organization of DDR protein accumulation at DNA DSBs. (A) DDR signal spreading. DDR proteins initially accumulate at DSB sites and then spread at distance via a positive feedback loop involving MDC1, which binds gH2AX, the MRN complex, and ATM kinase, which phosphorylates additional H2AX molecules further away from the break site. (B) Regional distribution of DDR proteins around DSBs. Factors involved in ATR signaling accumulate proximal to the break site on ssDNA generated by DNA end resection, while ATM signaling factors localize on flanking chromatin regions
 DDR signal spreading. DDR proteins initially accumulate at DSB sites and then spread at distance via a positive. feedback loop involving MDC1, which binds gH2AX, the MRN complex, and ATM kinase, which phosphorylates additional H2AX molecules further away from the break site. (B) Regional. distribution of DDR proteins around DSBs. Factors involved in ATR signaling accumulate proximal to the break site on ssDNA generated by DNA end resection, while ATM signaling factors. localize on flanking chromatin regions.")
11
Temporal regulation of DDR protein accumulation at DNA breaks
Temporal regulation of DDR protein accumulation at DNA breaks. (A) Sequential recruitment of DDR factors to SSBs and DSBs generated by laser microirradiation. (B) Cell cycle regulation of DDR foci formation. (Solid line) Efficient focus formation; (dashed line) weak/undetectable foci.
 Sequential recruitment of DDR factors to SSBs and DSBs generated by laser microirradiation. (B) Cell cycle regulation of DDR foci formation. (Solid line) Efficient focus formation; (dashed line) weak/undetectable foci.")
12
Proteine piattaforma SSB kinase signaling
Mre, Rad50 SSB Nbs1, a subunit of a complex that recognizes DNA DSBs HRR kinase signaling Binding platforms at DNA breaks. NBS1, MDC1, XRCC1, and XRCC4 act as binding platforms for the recruitment of other DDR factors to DNA breaks promoting DNA damage signaling and/or repair. Dotted lines indicate protein–protein interactions, while horizontal lines at the end of the dotted lines indicate interacting regions. Some interactions involve posttranslational modifications. (P) Phosphorylation; (PAR) PARylation. The red and the green semicircles represent BRCT and FHA domains, respectively. (b) Basic region at the end of the first BRCT domain of XRCC1 that interacts with poly(ADPribosyl) ated PARPs. DNA damage is a key factor both in the evolution and treatment of cancer. Genomic instability is a common feature of cancer cells, fuelling accumulation of oncogenic mutations, while radiation and diverse genotoxic agents remain important, if imperfect, therapeutic modalities. Cellular responses to DNA damage are coordinated primarily by two distinct kinase signaling cascades, the ATM-Chk2 and ATR-Chk1 pathways, which are activated by DNA double-strand breaks (DSBs) and single-stranded DNA respectively. Historically, these pathways were thought to act in parallel with overlapping functions; however, more recently it has become apparent that their relationship is more complex. In response to DSBs, ATM is required both for ATR-Chk1 activation and to initiate DNA repair via homologous recombination (HRR) by promoting formation of single-stranded DNA at sites of damage through nucleolytic resection. Interestingly, cells and organisms survive with mutations in ATM or other components required for HRR, such as BRCA1 and BRCA2, but at the cost of genomic instability and cancer predisposition. By contrast, the ATR-Chk1 pathway is the principal direct effector of the DNA damage and replication checkpoints and, as such, is essential for the survival of many, although not all, cell types. Remarkably, deficiency for HRR in BRCA1- and BRCA2-deficient tumors confers sensitivity to cisplatin and inhibitors of poly(ADP-ribose) polymerase (PARP), an enzyme required for repair of endogenous DNA damage. In addition, suppressing DNA damage and replication checkpoint responses by inhibiting Chk1 can enhance tumor cell killing by diverse genotoxic agents. Here, we review current understanding of the organization and functions of the ATM-Chk2 and ATR-Chk1 pathways and the prospects for targeting DNA damage signaling processes for therapeutic purposes. APTX is the gene involved in ataxia with oculomotor apraxia type 1 (AOA1), a recessive disorder with early-onset cerebellar ataxia, oculomotor apraxia and peripheral neuropathy. The encoded protein, aprataxin, is a DNA repair protein processing the products of abortive ligations, 5'-adenylated DNA. SSB
 Phosphorylation; (PAR) PARylation. The red and the green semicircles represent BRCT and FHA domains, respectively. (b) Basic region at the end of the first BRCT domain of XRCC1 that interacts with poly(ADPribosyl) ated PARPs. DNA damage is a key factor both in the evolution and treatment of cancer. Genomic instability is a common feature of cancer cells, fuelling accumulation of oncogenic mutations, while radiation and diverse genotoxic agents remain important, if imperfect, therapeutic modalities. Cellular responses to DNA damage are coordinated primarily by two distinct kinase signaling cascades, the ATM-Chk2 and ATR-Chk1 pathways, which are activated by DNA double-strand breaks (DSBs) and single-stranded DNA respectively. Historically, these pathways were thought to act in parallel with overlapping functions; however, more recently it has become apparent that their relationship is more complex. In response to DSBs, ATM is required both for ATR-Chk1 activation and to initiate DNA repair via homologous recombination (HRR) by promoting formation of single-stranded DNA at sites of damage through nucleolytic resection. Interestingly, cells and organisms survive with mutations in ATM or other components required for HRR, such as BRCA1 and BRCA2, but at the cost of genomic instability and cancer predisposition. By contrast, the ATR-Chk1 pathway is the principal direct effector of the DNA damage and replication checkpoints and, as such, is essential for the survival of many, although not all, cell types. Remarkably, deficiency for HRR in BRCA1- and BRCA2-deficient tumors confers sensitivity to cisplatin and inhibitors of poly(ADP-ribose) polymerase (PARP), an enzyme required for repair of endogenous DNA damage. In addition, suppressing DNA damage and replication checkpoint responses by inhibiting Chk1 can enhance tumor cell killing by diverse genotoxic agents. Here, we review current understanding of the organization and functions of the ATM-Chk2 and ATR-Chk1 pathways and the prospects for targeting DNA damage signaling processes for therapeutic purposes. APTX is the gene involved in ataxia with oculomotor apraxia type 1 (AOA1), a recessive disorder with early-onset cerebellar ataxia, oculomotor apraxia and peripheral neuropathy. The encoded protein, aprataxin, is a DNA repair protein processing the products of abortive ligations, 5 -adenylated DNA. SSB.")
13
The MDC1 TQXF motifs are ATM targets
P ATM The MDC1 TQXF motifs are ATM targets required for 53BP1 IRIF. (A) Domain architecture of MDC1, with ATM consensus sites (dots).
 Domain architecture. of MDC1, with ATM consensus sites (dots).")
14
Specialized binding modules for recognition of post-translational modifications (PTMs) at DNA breaks. The recruitment of DDR proteins to modified histones or othermodified proteins at sites of DNA breaks ismediated by specific interactions between the post-translational modification and a dedicated binding module. BRCTand FHA domains, which are represented by red and green semicircles, bind phosphorylated serine or threonine residues; Tudor domains, chromodomains, and PDH finger domains bind methylated histones; bromodomains (Bromo) bind acetylated histones; and UBDs bind ubiquitylated proteins. The PAR-binding domain can take the form of a basic stretch of amino acids (Basic), a PARbinding zinc finger (PBZ), or a macrodomain (Macro). Note that some of these modules are found as tandem domains and that not all post-translational modifications are damage-induced (asterisk [*] denotes constitutive modifications). The species of the proteins are indicated, unless only human proteins are listed. (H.s.) Homo sapiens; (S.c.) S. cerevisiae, (S.p.) S. pombe. Polo and Jackson 418 GENES
 bind acetylated histones; and UBDs bind ubiquitylated. proteins. The PAR-binding domain can take the. form of a basic stretch of amino acids (Basic), a PARbinding. zinc finger (PBZ), or a macrodomain (Macro). Note that some of these modules are found as tandem. domains and that not all post-translational modifications. are damage-induced (asterisk [*] denotes constitutive. modifications). The species of the proteins are. indicated, unless only human proteins are listed. (H.s.) Homo sapiens; (S.c.) S. cerevisiae, (S.p.) S. pombe. Polo and Jackson. 418 GENES.")
15
Domain architecture of RNF8
Ubiquitin ligase activity Forkhead associated(FHA) domain bind phosphothreonine-bearing epitopes interaction with ATM-phosphorylated MDC1. FHA-(R42A) and RING finger (C406s) mutants. Forkhead domain
 domain. bind phosphothreonine-bearing epitopes interaction with ATM-phosphorylated MDC1. FHA-(R42A) and. RING finger (C406s) mutants. Forkhead domain.")
16
Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase (Nadine Science Feb2008)
Cells respond to DSBs by recruiting the DNA-damage mediator protein MDC1, the p53-binding protein 1 (53BP1), and the breast cancer susceptibility protein BRCA1 to sites of damaged DNA.
, and the breast cancer susceptibility protein BRCA1 to sites of damaged DNA.")
17
Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase (Nadine Science Feb2008)
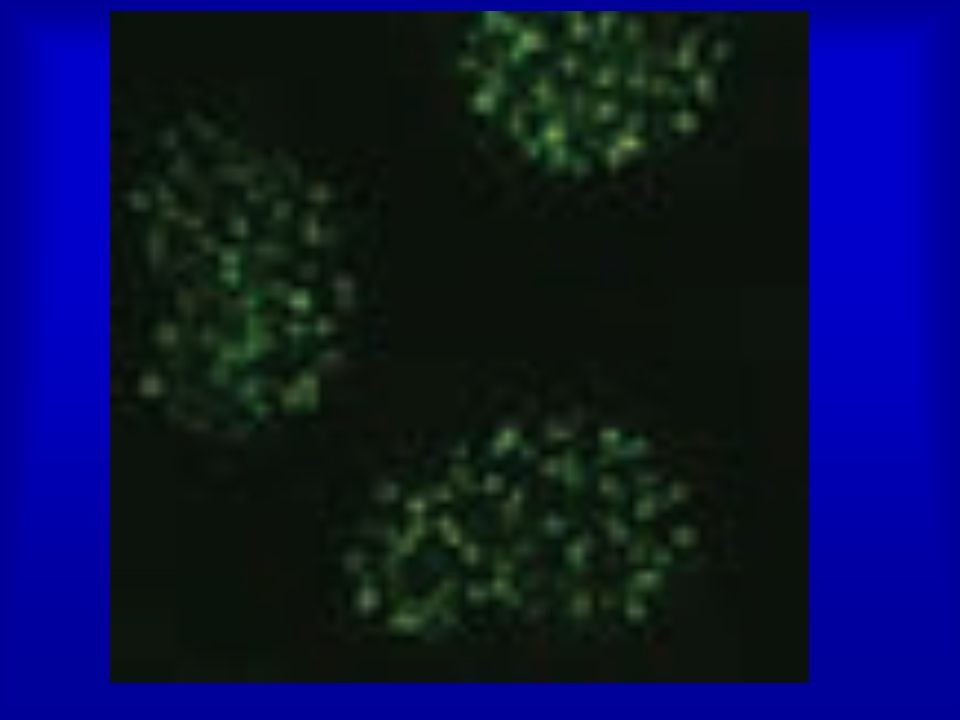
Cells respond to DSBs by recruiting the DNA-damage mediator protein MDC1, the p53-binding protein 1 (53BP1) to sites of damaged DNA. 53BP1 is an established player- important role in modulating chromatin structure surrounding the break site- in the cellular response to DNA damage and is a canonical component of ionizing-radiation induced foci (IRIF) - that cadre of proteins which assemble at DNA double strand breaks following radiation exposure and which are readily visualized by immunofluorescence microscopy. While its roles in p53 regulation and cell cycle checkpoint activation have been studied for some time, the impact of 53BP1 on DNA double strand break rejoining has only come to light in the past few years. Convincing evidence now exists for 53BP1 significantly affecting the outcome of DNA double strand break repair in several contexts, many of which hint to an important role in modulating chromatin structure surrounding the break site.
 to sites of damaged DNA. 53BP1 is an established player- important role in modulating chromatin structure surrounding the break site- in the cellular response to DNA damage and is a canonical component of ionizing-radiation induced foci (IRIF) - that cadre of proteins which assemble at DNA double strand breaks following radiation exposure and which are readily visualized by immunofluorescence microscopy. While its roles in p53 regulation and cell cycle checkpoint activation have been studied for some time, the impact of 53BP1 on DNA double strand break rejoining has only come to light in the past few years. Convincing evidence now exists for 53BP1 significantly affecting the outcome of DNA double strand break repair in several contexts, many of which hint to an important role in modulating chromatin structure surrounding the break site.")
20
Ranking by z score of 500 siRNAs giving the least
53BP1 foci from a siRNA screen

23
Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase (Nadine Science Feb2008)
Cells respond to DSBs by recruiting the DNA-damage mediator protein MDC1, the p53-binding protein 1 (53BP1), and the breast cancer susceptibility protein BRCA1 to sites of damaged DNA.
, and the breast cancer susceptibility protein BRCA1 to sites of damaged DNA.")
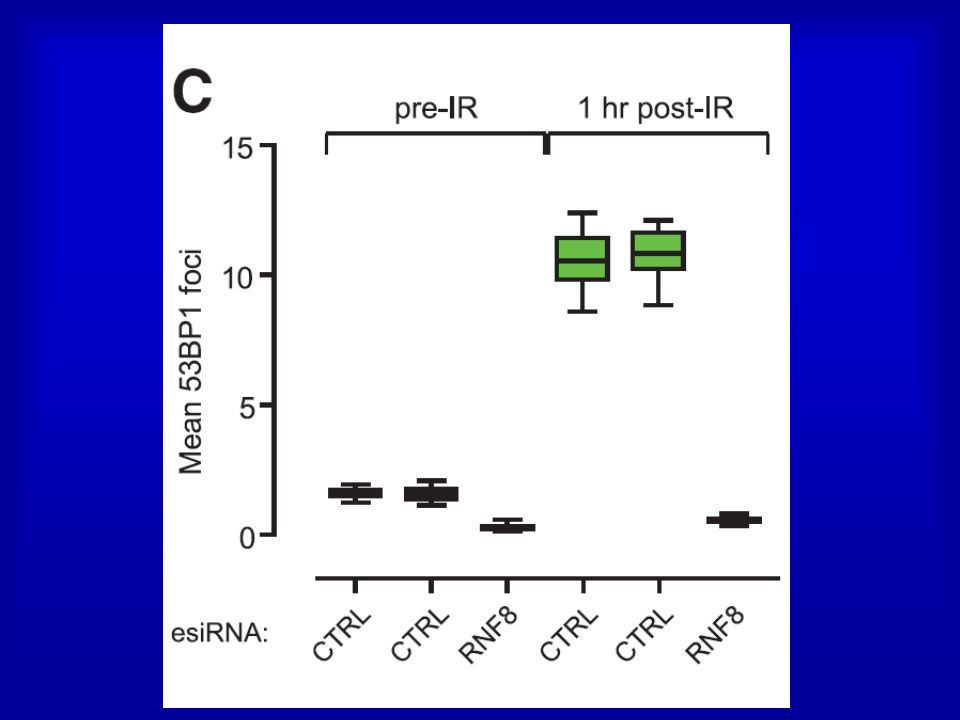
24
Irradiated (10 Gy) HeLa cells transfected with the indicated
siRNAs were stained with antibodies to gH2AX, BRCA1

26
Orchestration of the DNA-Damage Response by the RNF8 Ubiquitin Ligase (Nadine Science Feb2008)
Cells respond to DSBs by recruiting the DNA-damage mediator protein MDC1, the p53-binding protein 1 (53BP1), and the breast cancer susceptibility protein BRCA1 to sites of damaged DNA. The ubiquitin ligase RNF8 mediates ubiquitin conjugation and 53BP1 and BRCA1 focal accumulation at sites of DNA lesions. MDC1 recruits RNF8 through phosphodependent interactions between the RNF8 forkhead-associated domain and motifs in MDC1 that are phosphorylated by the DNA-damage activated protein kinase ataxia telangiectasia mutated (ATM). Depletion of the E2 enzyme UBC13 impairs 53BP1 recruitment to sites of damage, which suggests that it cooperates with RNF8. RNF8 promotes the G2/M DNA damage checkpoint and resistance to ionizing radiation. the DNA-damage response is orchestrated by ATM-dependent phosphorylation of MDC1 and RNF8-mediated ubiquitination.
, and the breast cancer susceptibility protein BRCA1 to sites of damaged DNA. The ubiquitin ligase RNF8 mediates ubiquitin conjugation and 53BP1 and BRCA1 focal accumulation at sites of DNA lesions. MDC1 recruits RNF8 through phosphodependent interactions between the RNF8 forkhead-associated domain and motifs in MDC1 that are phosphorylated by the DNA-damage activated protein kinase ataxia telangiectasia mutated (ATM). Depletion of the E2 enzyme UBC13 impairs 53BP1 recruitment to sites of damage, which suggests that it cooperates with RNF8. RNF8 promotes the G2/M DNA damage checkpoint and resistance to ionizing radiation. the DNA-damage response is orchestrated by ATM-dependent phosphorylation of MDC1 and RNF8-mediated ubiquitination.")
27
Mutazioni in BRCA 1 o 2 inattivazione meccanismo HRR predisposizione allo sviluppo di Carcinoma mammario ereditario, con insorgenza precoce tumore seno e ovaie BRCA 1: 50% mutazioni tumore mammario famigliare BRCA 2: 35% mutazioni tumore mammario famigliare Eredità di un allele mutante predisposizione al tumore, che insorge solo quando la seconda copia del gene è persa o mutata (perdita di eterozigosità)
")
28
BRCA 1 impairment of homologous repair in Brca1-deficient mouse embryonic stem cells increase in the frequency of NHEJ in Brca1-deficient cells

29
Riparazione per ricombinazione omologa (HRR)
Ripara le DSBs (Double-Strand Breaks) causate da radiazioni, stress ossidativo, farmaci Replicazione e trascrizione vengono bloccate nel sito della DSB e le estremità esposte sono soggette a degradazione con perdita di materiale genetico importanza HRR HRR utilizza come stampo il cromatidio fratello protezione dagli errori HRR avviene in tarda fase S o in G2, quando i cromatidi fratelli sono vicini
 causate da radiazioni, stress ossidativo, farmaci. Replicazione e trascrizione vengono bloccate nel sito della DSB e le estremità esposte sono soggette a degradazione con perdita di materiale genetico importanza HRR. HRR utilizza come stampo il cromatidio fratello protezione dagli errori. HRR avviene in tarda fase S o in G2, quando i cromatidi fratelli sono vicini.")
30
(ss DNA replication binding protein)
RPA (ss DNA replication binding protein) la polimerizzazione di RAD51 sul 3’ libero è BRCA1/2-dipendente Sensor MRN RAD51 (aiutata dall’elicasi RAD54) cerca la sequenza omologa sul cromatidio fratello e invade la doppia elica; le regioni 3’ a singola elica si appaiano con quelle complementari
 la polimerizzazione di RAD51 sul 3’ libero è BRCA1/2-dipendente. Sensor. MRN. RAD51 (aiutata dall’elicasi RAD54) cerca la sequenza omologa sul cromatidio fratello e invade la doppia elica; le regioni 3’ a singola elica si appaiano con quelle complementari.")
31
La DNA polimerasi allunga l’estremità 3’ libera (in cui si trova la nucleoproteina RAD51) utilizzando come stampo il cromatidio omologo non danneggiato L’altra regione 3’ a singola elica attorno alla zona danneggiata si appaia all’elica ormai corretta e gli eventuali gap sono riempiti da polimerasi e ligasi

32
BRCA1/BRCA2 PALB2 and BRCA1 are co-mediators that directly and indirectly, respectively, interact with BRCA2 and assist in localizing it, with RAD51 bound, to the sites of DNA damage Simplified schematic to depict the mediator step of recombination: displacement of RPA by RAD51. A critical step in homologous re-combinational repair is the displacement of RPA, the trimeric ssDNA-binding protein, by the RAD51 strand transfer protein. This step is highly regulated by cells to ensure that potentially deleterious events are avoided. Many different proteins are involved in assisting RAD51 to displace RPA at this stage, and the proteins that are directly involved are known as the ‘recombination mediators’ (Table 1). There are also additional proteins that function to assist the mediators or assist in their localization to DNA damage, and in this review, the proteins assisting the mediators are defined as ‘recombination co-mediators’. Human BRCA2 is a mediator that interacts directly with approximately eight RAD51 molecules and transports them to the site of ss-DNA bound by RPA, while PALB2 and BRCA1 are co-mediators that directly and indirectly, respectively, interact with BRCA2 and assist in localizing it, with RAD51 bound, to the sites of DNA damage (for references, see Table 1). DSB, double-strand DNA break; M/R/N, MRE11-RAD50-NBS1 complex. NAR 2010 Human BRCA2 is a mediator that interacts directly with approximately eight RAD51 molecules and transports them to the site of ss-DNA bound by RPA
. There are also additional proteins that function to assist the mediators or assist in their localization to DNA damage, and in this review, the proteins assisting the mediators are defined as ‘recombination co-mediators’. Human BRCA2 is a mediator that interacts directly with approximately eight RAD51 molecules and transports them to the site of ss-DNA bound by RPA, while PALB2 and BRCA1 are co-mediators that directly and indirectly, respectively, interact with BRCA2 and assist in localizing it, with RAD51 bound, to the sites of DNA damage (for references, see Table 1). DSB, double-strand DNA break; M/R/N, MRE11-RAD50-NBS1 complex. NAR Human BRCA2 is a mediator that interacts directly with approximately eight RAD51 molecules and transports them to the site of ss-DNA bound by RPA.")
34
Determination of regions required for the BRCA1-PALB2 interaction.
(A) Graphical projection of association between PALB2 (residues 9 – 42) and BRCA1 (residues 1393–1424) coiled-coil domains. Positions of the heptad repeat (positions a to g) were predicted by the Coil program Boxed residues were experimentally demonstrated to be responsible for the hetero-oligomeric interaction between PALB2 and BRCA1. Mutations in breast cancer susceptibility gene 1 and 2 (BRCA1 and BRCA2) predispose individuals to breast and ovarian cancer development. We previously reported an in vivo interaction between BRCA1 and BRCA2. However, the biological significance of their association is thus far undefined. Here, we report that PALB2, the partner and localizer of BRCA2, binds directly to BRCA1, and serves as the molecular scaffold in the formation of the BRCA1-PALB2- BRCA2 complex. The association between BRCA1 and PALB2 is primarily mediated via apolar bonding between their respective coiled-coil domains. More importantly, BRCA1 mutations identified in cancer patients disrupted the specific interaction between BRCA1 and PALB2. Consistent with the converging functions of the BRCA proteins in DNA repair, cells harboring mutations with abrogated BRCA1-PALB2 interaction resulted in defective homologous recombination (HR) repair. We propose that, via its direct interaction with PALB2, BRCA1 fine-tunes recombinational repair partly through its modulatory role in the PALB2-dependent loading of BRCA2-RAD51 repair machinery at DNA breaks. Our findings uncover PALB2 as the molecular adaptor between the BRCA proteins, and suggest that impaired HR repair is one of the fundamental causes for genomic instability and tumorigenesis observed in patients carrying BRCA1, BRCA2, or PALB2 mutations.
 Graphical projection of. association between PALB2 (residues 9 – 42) and BRCA1 (residues 1393–1424) coiled-coil domains. Positions of the heptad repeat (positions a to g) were predicted by the Coil program. Boxed residues were experimentally demonstrated to be responsible for the hetero-oligomeric interaction between PALB2 and BRCA1. Mutations in breast cancer susceptibility gene 1 and 2 (BRCA1 and. BRCA2) predispose individuals to breast and ovarian cancer development. We previously reported an in vivo interaction between. BRCA1 and BRCA2. However, the biological significance of their. association is thus far undefined. Here, we report that PALB2, the. partner and localizer of BRCA2, binds directly to BRCA1, and serves. as the molecular scaffold in the formation of the BRCA1-PALB2- BRCA2 complex. The association between BRCA1 and PALB2 is. primarily mediated via apolar bonding between their respective. coiled-coil domains. More importantly, BRCA1 mutations identified. in cancer patients disrupted the specific interaction between. BRCA1 and PALB2. Consistent with the converging functions of the. BRCA proteins in DNA repair, cells harboring mutations with. abrogated BRCA1-PALB2 interaction resulted in defective homologous. recombination (HR) repair. We propose that, via its direct. interaction with PALB2, BRCA1 fine-tunes recombinational repair. partly through its modulatory role in the PALB2-dependent loading. of BRCA2-RAD51 repair machinery at DNA breaks. Our findings. uncover PALB2 as the molecular adaptor between the BRCA proteins, and suggest that impaired HR repair is one of the fundamental. causes for genomic instability and tumorigenesis observed. in patients carrying BRCA1, BRCA2, or PALB2 mutations.")
36
BRCA 1 The BRCA1 tumor suppressor exists as a heterodimeric complex with BARD1, and this complex is thought to mediate many of the functions ascribed to BRCA1, including its role in tumor suppression. The two proteins share a common structural organization : an N-terminal RING domain and two C-terminal BRCT motifs the BRCA1/BARD1 heterodimer enhances chromosome stability by promoting homology-directed repair (HDR) of double strand DNA breaks.
 of double strand DNA breaks.")
37
BRCA 1 - Gene oncosoppressore, localizzato sul cromosoma 17
Espressione nel nucleo di molti tipi cellulari N-ter: dominio RING FINGER, coordina il legame con 2 ioni zinco contatta DNA C-ter: dominio BRCT induce / reprime geni; interazione con RNA pol II

38
BRCA 1 Ubc13 and Rnf8are recruited to sites of DNA damage through DNA-damage-induced phosphorylation of a chromatin-associated protein The Brca1 A complex contains Brca1/Bard1, Abraxas, Rap80, and Brcc36; however, with the exception of the Brca1–Abraxas interaction, how the A complex is assembled is not known. The A complex is localized to sites of DNA damage through the UIM domains of RAP80, which bind K63-linked polyubiquitin chains. In this study, we identified an FHA domain RING finger E3 ubiquitin ligase, RNF8, and an E2-conjugating enzyme known to form K63–polyubiquitin chains, Ubc13, each of which is required to recruit the Brca1Acomplex to sites of DNA damage. Rnf8 localizes to sites of DNA damage through an FHA-domain-containing region. We found that Rap80 contains an Abraxas interaction domain [AIR (Abraxas-interacting region)], required for association of Rap80 with Abraxas, Brca1, and Brcc36. Abraxas and Brcc36 associate through coiled-coil domains on each protein. These data suggest a model through which Ubc13 and Rnf8 are recruited to sites of DNA damage through DNA-damage-induced phosphorylation of a chromatin-associated protein and generate polyubiquitin chains that then recruit Rap80 and the entire Brca1 A complex to DNA-damage foci. This sequential E3 ubiquitin ligase recruitment constitutes a ubiquitin ligase cascade required for DNA repair and checkpoint signaling Ubc13 and Rnf8are recruited to sites of DNA damage through DNA-damage-induced phosphorylation of a chromatin-associated protein
], required. for association of Rap80 with Abraxas, Brca1, and Brcc36. Abraxas and Brcc36 associate through coiled-coil domains on each protein. These data suggest a model through which Ubc13 and Rnf8 are recruited to sites of DNA damage through DNA-damage-induced phosphorylation of a chromatin-associated protein and generate polyubiquitin chains that then recruit Rap80 and the entire Brca1 A complex to DNA-damage foci. This sequential E3 ubiquitin ligase recruitment constitutes a ubiquitin ligase cascade required for DNA repair and checkpoint signaling. Ubc13 and Rnf8are recruited to sites of DNA damage through DNA-damage-induced phosphorylation of a chromatin-associated protein.")
39
BRCA 1 An E3 ubiquitin ligase mediates the transfer of activated ubiquitin from an E2 ubiquitin-conjugating enzyme to its substrate lysine residues. BRCA1 has the ability to direct the synthesis of specific polyubiquitin chain linkages, depending on the E2 bound to its RING.

40
Regolazione delle proliferazione cellulare
Lega p53 attivando p21 Regolazione delle proliferazione cellulare Attiva TNFa Lega Rad50/51 e BRCA2 Riparazione HRR Inibisce la trasduzione del segnale da parte del recettore per l’estrogeno Reprime la proliferazione dell’epitelio mammario che è estrogeno-dipendente Mutazioni in BRCA1 possono determinare un’incontrollata proliferazione, in particolare delle cellule che rispondono all’estrogeno tumore Una volta attivata, p53 trascrive molti geni incluso quello per la p21, la quale lega i complessi G1-S/CDK e S/CDK (molecole molto importanti per la transizione dalla fase G1 alla fase S) inibendo la loro attività (ed evitando così la proliferazione della cellula mutata).
 inibendo la loro attività (ed evitando così la proliferazione della cellula mutata).")
41
BRCA1 regulates human mammary stem/progenitor cell fate
PNAS February 5, 2008

42
BRCA1 is implicated in multiple cellular functions
Red, ubiquitylation; Blue, transcription; Green, cell cyclecheckpoint control; Light blue, DNA repair; Gray, chromatin modifications.

43
Riparazione per ricombinazione non omologa (NHEJ)
E’ il meccanismo prevalente per la riparazione delle DSBs, soprattutto negli organismi con elevato numero di sequenze ripetute identiche (l’HRR rischierebbe di appaiare sequenze ripetute di cromosomi diversi) Può causare l’introduzione di errori nel codice genetico (perdita o aggiunta di nucleotidi) La via nota è quella Ku-dipendente, ma il NHEJ può avvenire, con meno efficienza, anche in estratti cellulari privi di Ku e DNA-PK esistenza di vie Ku-indipendenti non note
 Può causare l’introduzione di errori nel codice genetico (perdita o aggiunta di nucleotidi) La via nota è quella Ku-dipendente, ma il NHEJ può avvenire, con meno efficienza, anche in estratti cellulari privi di Ku e DNA-PK esistenza di vie Ku-indipendenti non note.")
44
In mammalian cells, most DSBs are preferentially repaired by NHEJ.
Mammals have evolved at least two genetically discrete ways to mediate DNA DSB repair: homologous recombination (HR) and non-homologous end joining (NHEJ). In mammalian cells, most DSBs are preferentially repaired by NHEJ. NHEJ consists of at least two sub-pathways— the main Ku heterodimer-dependent or “classic” NHEJ (C-NHEJ) pathway and an “alternative” NHEJ (A-NHEJ) pathway, which usually generates microhomology-mediated signatures at repair junctions. One of the most harmful lesions a cell can encounter is a DNA double-strand break (DSB). In all organisms, efficient repair of these DSBs is critical for the maintenance of genomic integrity and viability [1]. Unfortunately, DSBs are frequently generated endogenously during normal cellular processes such as DNA replication, lymphoid V(D)J or class-switch recombination and are induced exogenously by the exposure to a variety of genotoxic agents such as ionizing radiation or chemotherapeutics [2]. Cells have conspired to meet this demand on their genetic material with the evolution of two mechanistically distinct pathways to repair DSBs: homologous recombination (HR), which takes advantage of either a homologous chromosome or a sister chromatid to join the broken DNA ends [3] and non-homologous end joining (NHEJ), a process that directly joins the DSB with little or no sequence homology between the broken ends [2]. In bacteria and lower eukaryotes, HR dominates the DNA DSB repair events whereas in higher eukaryotes, and especially in mammals, NHEJ is the preferred pathway for DNA DSB repair. NHEJ consists of at least two genetically and biochemically distinct sub-pathways: a main—“classic”—end-joining pathway (C-NHEJ) and one interchangeably referred to as microhomology-mediated end joining (MMEJ) [4], alternative NHEJ (A-NHEJ), or backup NHEJ (B-NHEJ) [5],[6] (hereafter referred to as A-NHEJ). C-NHEJ, while by no means precise, results in minimal DNA end processing, whereas A-NHEJ mechanistically results in deletions per force that are often accompanied by microhomology at the repair junction {[7],[8]; reviewed by [6],[9]}. There are at least seven proteins required for C-NHEJ: Ku70, Ku86, the DNA dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, X-ray cross complementing 4 (XRCC4), XRCC4-like factor (XLF) and DNA ligase IV (LIGIV) {reviewed by [10]}. The basic mechanism of C-NHEJ has been worked out in great detail. Ku70 and Ku86 form a heterodimer (Ku) that contains an internal cavity, which Ku uses to bind to and encircle broken DNA ends [11]. Ku, besides protecting DNA ends from exonucleolytic attack, also recruits DNA-PKcs, a phosphoinositol-3-like family serine/threonine protein kinase [12]. Together, Ku70, Ku86 and DNA-PKcs form the DNA-dependent protein kinase complex (DNA-PK) and the assembly of this trimeric complex on the ends of double-stranded DNA activates the kinase activity of DNA-PKcs. DNA-PKcs, in turn, phosphorylates and activates the nuclease Artemis, which facilitates “cleaning up” of the ends. As a final step, ligation of the broken ends is catalyzed by the trimeric LIGIV complex, which consists of the catalytic core, DNA LIGIV, and its two accessory factors, XLF and XRCC4. In contrast to C-NHEJ, the mechanism, the regulation and the factors involved in A-NHEJ remain elusive. Mechanistically, it is believed that during A-NHEJ both broken ends are resected 5′-to-3′ on one strand to generate 3′-single-stranded overhangs containing regions of microhomology (generally a few nucleotides), which are then used to mediate the repair event. Because of this reaction pathway, deletion of the sequences between the microhomologies occurs as does deletion of one of the blocks of (micro)homology. Moreover, the remaining block of microhomology always resides at the precise site of repair and can be used as a landmark to define such repair events [6],[9]. A-NHEJ was not thought to be a very robust nor particularly important DSB repair pathway because it could usually only be detected in the absence of C-NHEJ. Indeed, one of the first descriptions of A-NHEJ came with the observation that the few NHEJ DSB repair events that could be detected in Ku86-deficient budding yeast occurred between short direct repeats [7]. Since then, there have similar reports in fission yeast [13], frogs [14] and several mammalian systems [15]–[22] including humans [23]. The significance of—and parallel interest in—A-NHEJ increased with the demonstration that A-NHEJ could substitute at reasonable levels for C-NHEJ during DNA DSB repair events in murine lymphoid class switch recombination [24],[25] and during certain types of aberrant V(D)J recombination reactions [26],[27]. Moreover, A-NHEJ has been implicated in the generation of large deletions and other genomic rearrangements in murine cells [28]–[30]. Similarly, microhomology has been found at the recombination junctions of radiation-induced genomic rearrangements [31],[32] implying that even radiation-induced DSBs can be repaired by A-NHEJ. Lastly, microhomologies are frequently detected at breakpoints for chromosomal deletions and translocations in human cancer cells [33],[34]. These observations have propelled many laboratories to identify the factors required for A-NHEJ. These studies have implicated poly (ADP-ribose) polymerase-1 (PARP-1), X-ray cross complementing 1 (XRCC1), DNA ligase III (LIGIII), polynucleotide kinase (PNK), Flap endonuclease 1 (Fen-1) [5], [14], [35]–[37] and most recently the Mre11Rad50Nbs1 (MRN) complex (reviewed by [38]) but it is clear that additional factors await identification. One of the most compelling questions in the DSB repair field is how pathway choice is determined. That is, once a chromosome breaks, how does the cell determine whether HR, C-NHEJ or A-NHEJ will mediate its repair? Since each of these repair pathways generates a discretely distinct product, the answer to this question is biologically important. Several laboratories have suggested that the relative abundance of factors, binding affinities for DNA ends, cell type specificity and/or cell cycle phases may impact upon this decision {reviewed in [39]}. These issues are complicated even more in human somatic cells where the impact of loss-of-function mutations on some of the C-NHEJ genes has distinctly different phenotypes than are observed in other mammals. In particular, Ku70 and Ku86 have evolved an essential telomere maintenance function that does not seem to be evident in any other mammalian studied to date [40]–[43]. Interestingly, Ku seems to exert this function by repressing the HR-mediated disassembly of telomeres [44] suggesting that pathway choice is critical for naturally occurring double-stranded DNA ends as well as broken ones.
 and non-homologous end joining (NHEJ). In mammalian cells, most DSBs are preferentially repaired by NHEJ. NHEJ consists of at least two sub-pathways— the main Ku heterodimer-dependent or classic NHEJ (C-NHEJ) pathway and. an alternative NHEJ (A-NHEJ) pathway, which usually generates microhomology-mediated signatures at repair junctions. One of the most harmful lesions a cell can encounter is a DNA double-strand break (DSB). In all organisms, efficient repair of these DSBs is critical for the maintenance of genomic integrity and viability [1]. Unfortunately, DSBs are frequently generated endogenously during normal cellular processes such as DNA replication, lymphoid V(D)J or class-switch recombination and are induced exogenously by the exposure to a variety of genotoxic agents such as ionizing radiation or chemotherapeutics [2]. Cells have conspired to meet this demand on their genetic material with the evolution of two mechanistically distinct pathways to repair DSBs: homologous recombination (HR), which takes advantage of either a homologous chromosome or a sister chromatid to join the broken DNA ends [3] and non-homologous end joining (NHEJ), a process that directly joins the DSB with little or no sequence homology between the broken ends [2]. In bacteria and lower eukaryotes, HR dominates the DNA DSB repair events whereas in higher eukaryotes, and especially in mammals, NHEJ is the preferred pathway for DNA DSB repair. NHEJ consists of at least two genetically and biochemically distinct sub-pathways: a main— classic —end-joining pathway (C-NHEJ) and one interchangeably referred to as microhomology-mediated end joining (MMEJ) [4], alternative NHEJ (A-NHEJ), or backup NHEJ (B-NHEJ) [5],[6] (hereafter referred to as A-NHEJ). C-NHEJ, while by no means precise, results in minimal DNA end processing, whereas A-NHEJ mechanistically results in deletions per force that are often accompanied by microhomology at the repair junction {[7],[8]; reviewed by [6],[9]}. There are at least seven proteins required for C-NHEJ: Ku70, Ku86, the DNA dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, X-ray cross complementing 4 (XRCC4), XRCC4-like factor (XLF) and DNA ligase IV (LIGIV) {reviewed by [10]}. The basic mechanism of C-NHEJ has been worked out in great detail. Ku70 and Ku86 form a heterodimer (Ku) that contains an internal cavity, which Ku uses to bind to and encircle broken DNA ends [11]. Ku, besides protecting DNA ends from exonucleolytic attack, also recruits DNA-PKcs, a phosphoinositol-3-like family serine/threonine protein kinase [12]. Together, Ku70, Ku86 and DNA-PKcs form the DNA-dependent protein kinase complex (DNA-PK) and the assembly of this trimeric complex on the ends of double-stranded DNA activates the kinase activity of DNA-PKcs. DNA-PKcs, in turn, phosphorylates and activates the nuclease Artemis, which facilitates cleaning up of the ends. As a final step, ligation of the broken ends is catalyzed by the trimeric LIGIV complex, which consists of the catalytic core, DNA LIGIV, and its two accessory factors, XLF and XRCC4. In contrast to C-NHEJ, the mechanism, the regulation and the factors involved in A-NHEJ remain elusive. Mechanistically, it is believed that during A-NHEJ both broken ends are resected 5′-to-3′ on one strand to generate 3′-single-stranded overhangs containing regions of microhomology (generally a few nucleotides), which are then used to mediate the repair event. Because of this reaction pathway, deletion of the sequences between the microhomologies occurs as does deletion of one of the blocks of (micro)homology. Moreover, the remaining block of microhomology always resides at the precise site of repair and can be used as a landmark to define such repair events [6],[9]. A-NHEJ was not thought to be a very robust nor particularly important DSB repair pathway because it could usually only be detected in the absence of C-NHEJ. Indeed, one of the first descriptions of A-NHEJ came with the observation that the few NHEJ DSB repair events that could be detected in Ku86-deficient budding yeast occurred between short direct repeats [7]. Since then, there have similar reports in fission yeast [13], frogs [14] and several mammalian systems [15]–[22] including humans [23]. The significance of—and parallel interest in—A-NHEJ increased with the demonstration that A-NHEJ could substitute at reasonable levels for C-NHEJ during DNA DSB repair events in murine lymphoid class switch recombination [24],[25] and during certain types of aberrant V(D)J recombination reactions [26],[27]. Moreover, A-NHEJ has been implicated in the generation of large deletions and other genomic rearrangements in murine cells [28]–[30]. Similarly, microhomology has been found at the recombination junctions of radiation-induced genomic rearrangements [31],[32] implying that even radiation-induced DSBs can be repaired by A-NHEJ. Lastly, microhomologies are frequently detected at breakpoints for chromosomal deletions and translocations in human cancer cells [33],[34]. These observations have propelled many laboratories to identify the factors required for A-NHEJ. These studies have implicated poly (ADP-ribose) polymerase-1 (PARP-1), X-ray cross complementing 1 (XRCC1), DNA ligase III (LIGIII), polynucleotide kinase (PNK), Flap endonuclease 1 (Fen-1) [5], [14], [35]–[37] and most recently the Mre11Rad50Nbs1 (MRN) complex (reviewed by [38]) but it is clear that additional factors await identification. One of the most compelling questions in the DSB repair field is how pathway choice is determined. That is, once a chromosome breaks, how does the cell determine whether HR, C-NHEJ or A-NHEJ will mediate its repair Since each of these repair pathways generates a discretely distinct product, the answer to this question is biologically important. Several laboratories have suggested that the relative abundance of factors, binding affinities for DNA ends, cell type specificity and/or cell cycle phases may impact upon this decision {reviewed in [39]}. These issues are complicated even more in human somatic cells where the impact of loss-of-function mutations on some of the C-NHEJ genes has distinctly different phenotypes than are observed in other mammals. In particular, Ku70 and Ku86 have evolved an essential telomere maintenance function that does not seem to be evident in any other mammalian studied to date [40]–[43]. Interestingly, Ku seems to exert this function by repressing the HR-mediated disassembly of telomeres [44] suggesting that pathway choice is critical for naturally occurring double-stranded DNA ends as well as broken ones.")
45
L’eterodimero Ku 70/80 lega le DSBs, in tal modo proteggendo le estremità libere dalla degradazione finchè la giunzione non è completa Ku recluta DNA-PK DNA-PK ha siti di legame sia per le DSBs sia per il DNA a doppia elica adiacente ad esse e promuove l’allineamento delle regioni che hanno microomologie Autophosphorylation of the DNA-PKcatalytic subunit (DNA-PKcs) after recruitment of DNA-PKcs to DSBs by Ku
 after recruitment of DNA-PKcs to DSBs by Ku.")
46
cornerstone of the NHEJ pathway
Ku translocates inwards DNA-PKcs interacts with the nuclease Artemis synaptic complex IR induces multiple forms of DNA damage including DSBs that contain non-ligatable end groups such as 3’-phosphate and 3’-phosphoglycolate groups (indicated by ). (B) The Ku heterodimer (orange) binds the ends of the DSB, tethering the ends together. Recruitment of Ku to the DSB occurs independently of other known NHEJ or DSB repair proteins, consistent with Ku acting as the cornerstone of the NHEJ pathway. (C) Ku translocates inwards, allowing recruitment of DNA-PKcs (blue) such that it binds the extreme termini of the break (D). Recruitment of DNA-PKcs to the DSB requires Ku but no other NHEJ or DSB repair factors. Two DNA-PK molecules (DNA-PKcs bound to DNA-bound Ku) interact to tether the DSB together in what has been termed a “synaptic complex”. This triggers autophosphorylation (yellow circles) of DNA-PKcs in trans (E), inducing a conformational change that causes release of the DNA ends and/or release of phosphorylated DNA-PKcs from the complex. Whether DNA-PKcs is released prior to recruitment of the X4-L4 complex (green) and it’s associated factors (F), or whether it remains part of a multi-protein complex until repair is completed (M) is not known. Inhibition of the protein kinase activity of DNA-PKcs (step E), prevents dissociation of DNA-PKcs (step F), blocking access of NHEJ or other repair factors to the DSB, resulting in radiation sensitivity. (G) A portion of the total cellular DNA-PKcs interacts with the nuclease Artemis (red), but if or when Artemis is released from the DNA-PKcs complex (H) is not known. (I) PNK (pink) interacts with XRCC4 suggesting that it is recruited to the break with the X4-L4 complex (green) (J). XLF (yellow) and DNA pol μ and λ (purple) interact with both X4-L4 and Ku, suggesting that they are recruited after or at the same time as X4-L4 is recruited to the Ku-DNA complex (K). Other processing enzymes such as WRN and APLF (shown in grey) may also be recruited through interactions with DNA-bound Ku, XRCC4 and/or the X4-L4 complex (L). The order of recruitment of processing factors may be flexible and depend on the precise type of DNA damage present at the DSB. Multiple protein-protein and protein-DNA interactions may stabilize the formation of the complex at the DSB as well as aid in retention of NHEJ factors at the break. Once the ends are processed, the X4-L4 complex ligates the ends, repairing the break. Ligation of incompatible DNA ends is aided by the regulatory factor, XLF. How the various factors are released after repair is unknown, however, it is possible that ubiquitylation and/or proteolysis may be involved (N). Reactions requiring or enhanced by the presence of DNA-bound Ku are shown in red.
. (B) The Ku heterodimer (orange) binds the ends of the DSB, tethering the ends together. Recruitment of Ku to the DSB occurs independently of other known NHEJ or DSB repair proteins, consistent with Ku acting as the cornerstone of the NHEJ pathway. (C) Ku translocates inwards, allowing recruitment of DNA-PKcs (blue) such that it binds the extreme termini of the break (D). Recruitment of DNA-PKcs to the DSB requires Ku but no other NHEJ or DSB repair factors. Two DNA-PK molecules (DNA-PKcs bound to DNA-bound Ku) interact to tether the DSB together in what has been termed a synaptic complex . This triggers autophosphorylation (yellow circles) of DNA-PKcs in trans (E), inducing a conformational change that causes release of the DNA ends and/or release of phosphorylated DNA-PKcs from the complex. Whether DNA-PKcs is released prior to recruitment of the X4-L4 complex (green) and it’s associated factors (F), or whether it remains part of a multi-protein complex until repair is completed (M) is not known. Inhibition of the protein kinase activity of DNA-PKcs (step E), prevents dissociation of DNA-PKcs (step F), blocking access of NHEJ or other repair factors to the DSB, resulting in radiation sensitivity. (G) A portion of the total cellular DNA-PKcs interacts with the nuclease Artemis (red), but if or when Artemis is released from the DNA-PKcs complex (H) is not known. (I) PNK (pink) interacts with XRCC4 suggesting that it is recruited to the break with the X4-L4 complex (green) (J). XLF (yellow) and DNA pol μ and λ (purple) interact with both X4-L4 and Ku, suggesting that they are recruited after or at the same time as X4-L4 is recruited to the Ku-DNA complex (K). Other processing enzymes such as WRN and APLF (shown in grey) may also be recruited through interactions with DNA-bound Ku, XRCC4 and/or the X4-L4 complex (L). The order of recruitment of processing factors may be flexible and depend on the precise type of DNA damage present at the DSB. Multiple protein-protein and protein-DNA interactions may stabilize the formation of the complex at the DSB as well as aid in retention of NHEJ factors at the break. Once the ends are processed, the X4-L4 complex ligates the ends, repairing the break. Ligation of incompatible DNA ends is aided by the regulatory factor, XLF. How the various factors are released after repair is unknown, however, it is possible that ubiquitylation and/or proteolysis may be involved (N). Reactions requiring or enhanced by the presence of DNA-bound Ku are shown in red.")
47
Dati sperimentali: Davydov et al, 2003
Analisi su omogenati estratti dal cervello di 39 pazienti affetti da Alzheimer (AD) e 7 soggetti sani di pari età (C) EMSA: electrophoretic mobility shift assay Utilizza una sonda radioattiva contenente la sequenza di legame per la proteina di interesse (in questo caso Ku) e incubata con estratti nucleari Principio: in un gel di PAA i complessi DNA-Proteine migrano più lentamente rispetto a DNA libero
 e 7 soggetti sani di pari età (C) EMSA: electrophoretic mobility shift assay. Utilizza una sonda radioattiva contenente la sequenza di legame per la proteina di interesse (in questo caso Ku) e incubata con estratti nucleari. Principio: in un gel di PAA i complessi DNA-Proteine migrano più lentamente rispetto a DNA libero.")
48
Capacità di legame di Ku al DNA
Supershift: per confermare che la proteina che si è legata è Ku-80, si aggiunge nell’EMSA un anticorpo specifico. L’anticorpo andrà a legare la proteina Ku e se questa è legata al DNA marcato, il legame dell’anticorpo causerà un ulteriore ritardo nella corsa eleettrofeoretica Sonda libera Sonda - Ku Sonda – Ku - anticorpo Controllo sano Paziente Alzheimer

49
Alterazione di una delle proteine coinvolte nel meccanismo di NHEJ può determinare invecchiamento precoce

50
Dati sperimentali: Vogel et al., 1999
Topi privi di entrambe le copie del gene per Ku-86 (Ku 86-/-) Vita media Ku 86 -/-: 3814 wks Vita media controlli: 9717 wks Età massima raggiunta da un topo Ku 86 -/-: 87 wks Età massima raggiunta da un topo di controllo: 127 wks Topi Ku 86-/-: insorgenza precoce di tumori, atrofia della pelle e dei follicoli, chiusura delle epifisi, curvatura della spina dorsale, assottigliamento della pelle Le linee tratteggiate rappresentano la sovrapposizione dell’andamento delle due curve. Topi DNA-PK-/-: non vanno incontro ad invecchiamento precoce, forse perché l’attività fosforilante di DNA-PK può essere svolta da altre chinasi presenti nella cellula
 Vita media Ku 86 -/-: 3814 wks. Vita media controlli: 9717 wks. Età massima raggiunta da un topo Ku 86 -/-: 87 wks. Età massima raggiunta da un topo di controllo: 127 wks. Topi Ku 86-/-: insorgenza precoce di tumori, atrofia della pelle e dei follicoli, chiusura delle epifisi, curvatura della spina dorsale, assottigliamento della pelle. Le linee tratteggiate rappresentano la sovrapposizione dell’andamento delle due curve. Topi DNA-PK-/-: non vanno incontro ad invecchiamento precoce, forse perché l’attività fosforilante di DNA-PK può essere svolta da altre chinasi presenti nella cellula.")
51
Alterazioni nel NHEJ possono determinare invecchiamento precoce anche nell’uomo:
Sindrome di Werner: vita media di 50 anni, alopecia, osteoporosi, arteriosclerosi, cataratte, atrofia della pelle,… E’ causata da mutazioni nella proteina WRN, una DNA elicasi/nucleasi. WRN ha un ruolo sia nel HRR sia nel NHEJ (interagisce con Ku) IPOTESI: nei soggetti sani, quando la ricombinazione omologa non è possibile, WRN lega Ku e facilita la riparazione della DSB mediante NHEJ Nei pazienti affetti, invece, la ricombinazione omologa diventa meno efficiente e anche l’alternativa offerta dal NHEJ non è più favorita dal legame tra WRN e Ku
 IPOTESI: nei soggetti sani, quando la ricombinazione omologa non è possibile, WRN lega Ku e facilita la riparazione della DSB mediante NHEJ. Nei pazienti affetti, invece, la ricombinazione omologa diventa meno efficiente e anche l’alternativa offerta dal NHEJ non è più favorita dal legame tra WRN e Ku.")
Presentazioni simili























 in piante, C. elegans, Drosophila>")
