Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
FARMACOCINETICA

2
DOSE ASSORBIMENTO ESTERNO (vie di introduzione) SEDE DI F. LIBERO
FARMACO LIBERO PLASMA F. LEGATO ALLE PROTEINE METABOLITI TESSUTI DI DEPOSITO F. LEGATO F. LIBERO BIOTRASFORMAZIONI SEDE DI F. LIBERO AZIONE (RECETTORI) F. LEGATO ESCREZIONE
 F. LEGATO. ESCREZIONE.")
3
Fasi della Farmacocinetica
ASSORBIMENTO DISTRIBUZIONE METABOLISMO ELIMINAZIONE Le Interazioni farmacocinetiche possono quindi verificarsi a livello dell’assorbimento, della distribuzione, del metabolismo e dell’eliminazione del farmaco. Le interazioni farmacocinetiche sono assai difficili da prevedere soprattutto per quanto riguarda gli aspetti quantitativi delle modifiche indotte o delle conseguenze cliniche correlate ad esse. Alcune interazioni determinano importanti effetti clinici mentre in altri casi l’esito dell’interazione è di scarsa rilevanza clinica.

4
PRINCIPALI PARAMETRI FARMACOCINETICI
Cmax: concentrazione massima Tmax: tempo per raggiungere la Cmax F%: biodisponibilità AUC: area sotto la curva Vd: volume di distribuzione Cl: clearance T½: tempo necessario perché la concentrazione plasmatica si riduca della metà

5
tempo Steady State = Cmax Cmin T1/2 Cmin Concentrazione
AUC Cmin Concentrazione Plasmatica (g/ml)
")
6
AUC Relazione esemplificativa tra concentrazione plasmatica e tempo, dopo una singola dose orale di un farmaco ipotetico. L'area al di sotto della curva concentrazione plasmatica-tempo è indicata dall'ombreggiatura.

7
BIODISPONIBILITA’ La biodisponibilità varia da soggetto a soggetto ed anche nello stesso soggetto in relazione a diverse situazioni Concentrazioni seriche di propranololo in 6 volontari sani dopo somministrazione orale di 80 mg a digiuno o non a digiuno (Clinical Pharmacokinetics, 3: , 1978.)
")
8
AUC per la via endovenosa
la frazione di farmaco somministrato che raggiunge immodificato il circolo sistemico AUC per la via in esame AUC per la via endovenosa F = AUC per via orale (50 cm2) AUC per via endovenosa (100 cm2) F = 0.5
 AUC per via endovenosa (100 cm2) F = 0.5.")
9
VOLUME APPARENTE DI DISTRIBUZIONE (Vd)
Rappresenta il volume in cui si distribuisce un farmaco assumendo che raggiunga nei compartimenti extraplasmatici la stessa concentrazione che ha nel plasma. Vd = Quantità di farmaco somministrato Concentrazione plasmatica In taluni casi il Vd è superiore ai reali volumi corporei se l’accumulo tissutale determina una riduzione della concentrazione plasmatica 9

10
Distribuzione dei f. nei liquidi interstiziali e cellulari
H2O corporea totale 0.6 l/Kg (uomo di 70 kg: 42 lt) pari al 60% peso corporeo (piccole molecole idrosolubili, antipirina, alcool etilico) Nel neonato: 80%; Nel vecchio: 55% H2O extracellulare: 0.2 l/Kg (12-14 lt) pari al 29% acqua totale (penicillina G, mannitolo) H2O plasmatica: 0.04 l/Kg (ca. 3 lt) pari al 7% acqua totale (sangue 0.08 l/Kg = 5.5 lt) (es. furosemide = 7.7 lt) H2O intracellulare: 27 lt (64% acqua totale) Tessuto deposito: f. liposolubili con distribuzione prevalente nel tessuto adiposo (20-50% peso corporeo) (antidepressivi tric., digossina)
 pari al 60% peso corporeo (piccole molecole idrosolubili, antipirina, alcool etilico) Nel neonato: 80%; Nel vecchio: 55% H2O extracellulare: 0.2 l/Kg (12-14 lt) pari al 29% acqua totale (penicillina G, mannitolo) H2O plasmatica: 0.04 l/Kg (ca. 3 lt) pari al 7% acqua totale (sangue 0.08 l/Kg = 5.5 lt) (es. furosemide = 7.7 lt) H2O intracellulare: 27 lt (64% acqua totale) Tessuto deposito: f. liposolubili con distribuzione prevalente nel tessuto adiposo (20-50% peso corporeo) (antidepressivi tric., digossina)")
11
Come è possibile se volume H2O totale pari a circa 45 L?
VOLUME APPARENTE di DISTRIBUZIONE Vd = Quantità di farmaco somministrato Concentrazione plasmatica Vd (l) = D (mg) C0 (mg/l) = 100 33 = 3 l Ma per C0 = 1 mg/l allora Vd = 100 L !!!!! Come è possibile se volume H2O totale pari a circa 45 L? Tessuto deposito: Vd > lt: f. liposolubili con distribuzione prevalente nel tessuto adiposo (20-50% peso corporeo) (antidepressivi tric., digossina)
 = D (mg) C0 (mg/l) = = 3 l. Ma per C0 = 1 mg/l allora Vd = 100 L !!!!! Come è possibile se volume H2O totale pari a circa 45 L Tessuto deposito: Vd > lt: f. liposolubili con distribuzione prevalente nel tessuto adiposo (20-50% peso corporeo) (antidepressivi tric., digossina)")
12
VOLUME APPARENTE DI DISTRIBUZIONE
Vd = Dose/ Conc. Plasma Più la concentrazione plasmatica di un farmaco è elevata rispetto alla dose iniziale, più il valore numerico del Vd sarà piccolo ad indicare che il farmaco ha un basso volume di distribuzione. Al contrario una bassa concentrazione plasmatica rispetto alla dose indicherà che il farmaco si è distribuito in altri distretti dell’organismo e sarà dotato di un alto volume di distribuzione.

13
tempo Steady State = Cmax Cmin T1/2 Cmin Concentrazione
AUC Cmin T1/2 Steady State Cmin Cmax tempo Concentrazione Plasmatica (g/ml)
")
14
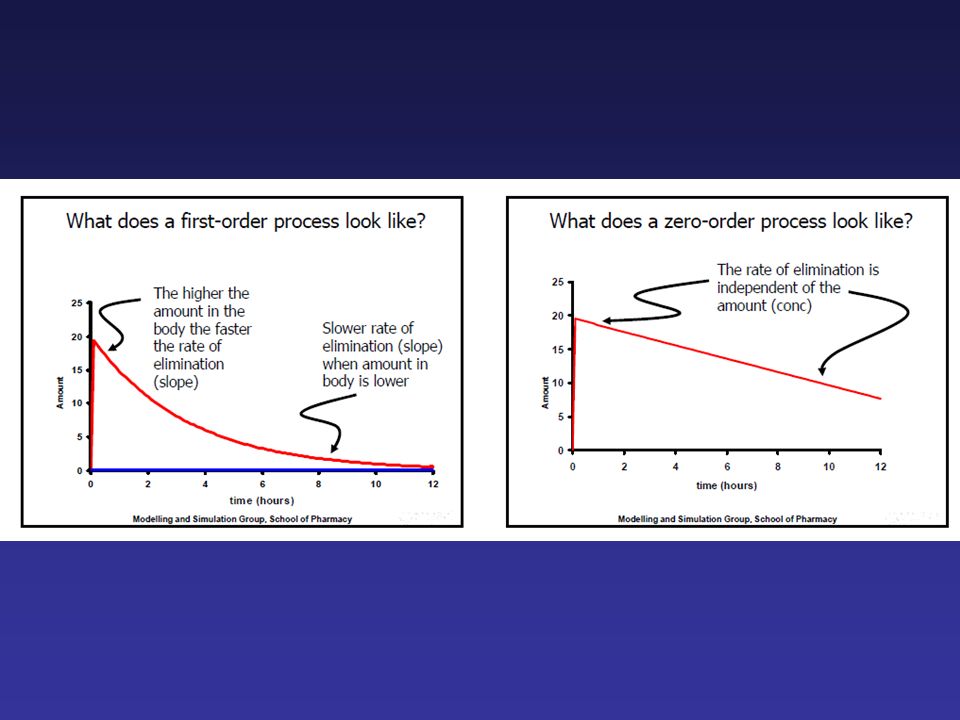
La farmacocinetica studia l’andamento della concentrazione plasmatica di un farmaco (C) nei liquidi corporei e nei tessuti in funzione del tempo (t). Cinetica di primo ordine: nell’unità di tempo viene eliminata (ma anche assorbita) una frazione costante della quantità di f. presente nell’organismo; la quantità assoluta di f. è proporzionale alla concentrazione plasmatica. I parametri farmacocinetici (Kel, T1/2, Cl) sono indipendenti dalla dose somministrata: cinetica dose-indipendente. Cinetica di ordine 0: nell’unità di tempo viene eliminata (ma anche assorbita) una quantità costante del f. presente nell’organismo: i parametri farmacocinetici dipendono dalla dose somministrata: cinetica dose-dipendente. Modelli compartimentali: organismo in compartimenti con un certo volume e comunicanti tra loro.
 una frazione costante della quantità di f. presente nell’organismo; la quantità assoluta di f. è proporzionale alla concentrazione plasmatica. I parametri farmacocinetici (Kel, T1/2, Cl) sono indipendenti dalla dose somministrata: cinetica dose-indipendente. Cinetica di ordine 0: nell’unità di tempo viene eliminata (ma anche assorbita) una quantità costante del f. presente nell’organismo: i parametri farmacocinetici dipendono dalla dose somministrata: cinetica dose-dipendente. Modelli compartimentali: organismo in compartimenti con un certo volume e comunicanti tra loro.")
16
Cinetica di I° ordine lnCt = lnC0 - ket Ct = C0·e-ket C0 = D/Vd
Concentrazione Plasmatica (g/ml) C0 = D/Vd lnCt = lnC0 - ket Concentrazione plasmatica (ln) A C1 Pendenza: -Ke Cinetica plasmatica di un composto dopo iniezione endovenosa e impianto dell’equazione della curva C0.5 B Ct = C0·e-ket t1/2 Tempo (ore) L'equazione indica che la concentrazione plasmatica del farmaco decade con un andamento monoesponenziale (B) secondo la costante ke. Se rappresentiamo graficamente in scala semilogaritmica le concentrazioni plasmatiche in funzione del tempo si ottiene una retta (A) che decade con una pendenza uguale a ke.
 C0 = D/Vd. lnCt = lnC0 - ket. Concentrazione plasmatica (ln) A. C1. Pendenza: -Ke. Cinetica plasmatica di un composto dopo iniezione endovenosa e impianto dell’equazione della curva. C0.5. B. Ct = C0·e-ket. t1/ Tempo (ore) L equazione indica che la concentrazione plasmatica del farmaco decade con un andamento monoesponenziale (B) secondo la costante ke. Se rappresentiamo graficamente in scala semilogaritmica le concentrazioni plasmatiche in funzione del tempo si ottiene una retta (A) che decade con una pendenza uguale a ke.")
17
Sono neccessarie 10 emivite per eliminare il 99,9%
Emivita Tempo (t1/2) necessario perché la concentrazione plasmatica del farmaco si dimezzi = Cmax AUC Cmin T1/2 N° di t½ Frazione di farmaco rimanente 1 2 3 4 5 6 7 8 9 10 100% 50% 25% 12.5% 6.25% 3.125% 1.56% 0.78% 0.39% 0.195% 0.0975% L'emivita si può calcolare matematicamente da Ct = C0∙e-ket L’emivita è un parametro che dipende dalla Clearance e dal Vd T1/2 = x Vd Cl Sono neccessarie 10 emivite per eliminare il 99,9%
 necessario perché la concentrazione plasmatica del farmaco si dimezzi. = Cmax. AUC. Cmin. T1/2. N° di t½. Frazione di farmaco rimanente % 50% 25% 12.5% 6.25% 3.125% 1.56% 0.78% 0.39% 0.195% % L emivita si può calcolare matematicamente da Ct = C0∙e-ket. L’emivita è un parametro che dipende dalla Clearance e dal Vd. T1/2 = x Vd. Cl. Sono neccessarie 10 emivite per eliminare il 99,9%")
18
Steady State Cmin Cmax tempo 194 187.5 175 150 97% 100 94% 87.5% 75% 50% Il tempo necessario per raggiungere la Css desiderata (Concentrazione bersaglio) è indipendente dalla dose ma dipende esclusivamente da t1/2: sono necessarie circa 4-5 emivite per raggiungere il 94-97% di Css. La conseguenza pratica di questo è che una Css terapeuticamente valida è raggiunta rapidamente con farmaci a emivita breve, e più lentamente con farmaci ad emivita lunga. Con quest'ultimo tipo di farmaci può essere necessario somministrare una dose d'attacco.
 è indipendente dalla dose ma dipende esclusivamente da t1/2: sono necessarie circa 4-5 emivite per raggiungere il 94-97% di Css. La conseguenza pratica di questo è che una Css terapeuticamente valida è raggiunta rapidamente con farmaci a emivita breve, e più lentamente con farmaci ad emivita lunga. Con quest ultimo tipo di farmaci può essere necessario somministrare una dose d attacco.")
19
varia in funzione del meccanismo di eliminazione coinvolto:
La Clearance renale varia in funzione del meccanismo di eliminazione coinvolto: farmaco filtrato e non riassorbito e secreto: Cl = 130 ml/min (inulina) farmaco filtrato e riassorbito, non secreto: Cl = < 130 ml/min Il glucosio viene completamente riassorbito (per glicemia 120 mg/ml) Cl = 0 farmaco completamente filtrato e secreto, non riassorbito: Cl = 650 ml/min (paraamminoippurico, penicillina)
 farmaco filtrato e riassorbito, non secreto: Cl = < 130 ml/min. Il glucosio viene completamente riassorbito (per glicemia 120 mg/ml) Cl = 0. farmaco completamente filtrato e secreto, non riassorbito: Cl = 650 ml/min (paraamminoippurico, penicillina)")
20
Velocità di eliminazione = Cltot x Cp
CLEARANCE SISTEMICA TOTALE: volume di plasma che viene completamente depurato dal farmaco nell'unità di tempo da tutti i meccanismi di eliminazione. Viene espressa come volume nell’unità di tempo (l/h/Kg): essa non dipende dalla dose somministrata. CLtot= velocità di eliminazione concentrazione Quantità escreta nell’unità di tempo = Cl x C Velocità di eliminazione = Cltot x Cp CLEARANCE RENALE Quantità (volume) di plasma che viene depurato dal farmaco nell’unità di tempo: U x V C x t Cl = Quantità escreta nell’unità di tempo = U x V t Quantità escreta nell’unità di tempo = Cl x C CLtot = Vd x 0,693/ t1/2
: essa non dipende dalla dose somministrata. CLtot= velocità di eliminazione. concentrazione. Quantità escreta nell’unità di tempo = Cl x C. Velocità di eliminazione = Cltot x Cp. CLEARANCE RENALE. Quantità (volume) di plasma che viene depurato dal farmaco nell’unità di tempo: U x V. C x t. Cl = Quantità escreta nell’unità di tempo = U x V. t. Quantità escreta nell’unità di tempo = Cl x C. CLtot = Vd x 0,693/ t1/2.")
Presentazioni simili





 + soluto In genere solvente liquido (es.acqua) E soluto solido, liquido, aeriforme.>")



1 VELOCITA DI REAZIONE ED EQUILIBRI.>")
2HPO4 è … 132 g 114 g>")








