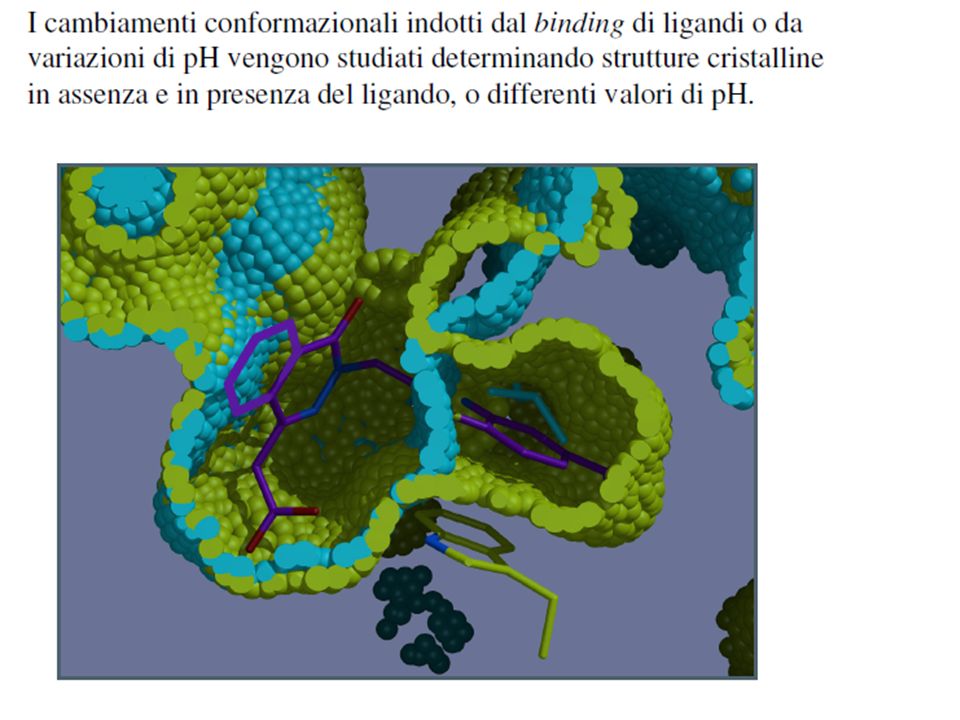
Scaricare la presentazione
1
Come si può studiare la struttura di una proteina
i metodi sperimentali classici per la risoluzione della struttura tridimensionale di una proteina sono: la cristallografia a raggi X la spettroscopia a risonanza magnetica e nucleare (Nuclear Magnetic Resonance, NMR)
")
2
diffrazione ai raggi X NMR formazione di cristalli cellula batterica
plasmide DNA esogeno formazione di cristalli NMR moltiplicazione del clone purificazione della proteina

4
Cristallografia a raggi X
La proteina, cristallizzata, viene bombardata con un raggio di fotoni collimati ad alta energia. I fotoni vengono diffratti in modo differente a seconda del tipo di atomo che colpiscono. I raggi diffratti vengono raccolti formando un quadro (pattern) di diffrazione Il pattern di diffrazione viene usato per ricostruire le coordinate dei singoli atomi che compongono la macromolecola e quindi la sua struttura 3D. I raggi X interagiscono quasi esclusivamente con gli elettroni presenti nella materia e non con i nuclei. Una struttura ai raggi X è quindi un’immagine della densità elettronica dell’oggetto in analisi
 di diffrazione. Il pattern di diffrazione viene usato per ricostruire le coordinate dei singoli atomi che compongono la macromolecola e quindi la sua struttura 3D. I raggi X interagiscono quasi esclusivamente con gli elettroni presenti nella materia e non con i nuclei. Una struttura ai raggi X è quindi un’immagine della densità elettronica dell’oggetto in analisi.")
5
Cristallografia a raggi X
Risoluzione ottenibile su piccole molecole organiche 1Å. In generale proteine cristallizate hanno grado di organizzazione più basso che limitano la risoluzione (2-3.5Å). Questo è dovuto anche all’alta idratazione dei cristalli (40-60% di acqua). Una tale risoluzione non è sufficiente per rivelare la posizione dei singoli atomi, ma è sufficiente per tracciare l’andamento e la disposizione dello scheletro covalente della proteina. Occorre quindi conoscere la sequenza primaria in modo da adattare la mappa della densità elettronica alla sequanza aminoacidica. Limiti del metodo: Cristallizzare proteine e` difficile Ricavare la struttura dal pattern di diffrazione e` computazionalmente complesso L’informazione che si ottiene e` statica, mentre la conforomazione di una proteina in soluzione varia nel tempo B factor (fattore di temperatura): indice “indiretto” di flessibilita’ strutturale
. Questo è dovuto anche all’alta idratazione dei cristalli (40-60% di acqua). Una tale risoluzione non è sufficiente per rivelare la posizione dei singoli atomi, ma è sufficiente per tracciare l’andamento e la disposizione dello scheletro covalente della proteina. Occorre quindi conoscere la sequenza primaria in modo da adattare la mappa della densità elettronica alla sequanza aminoacidica. Limiti del metodo: Cristallizzare proteine e` difficile. Ricavare la struttura dal pattern di diffrazione e` computazionalmente complesso. L’informazione che si ottiene e` statica, mentre la conforomazione di una proteina in soluzione varia nel tempo. B factor (fattore di temperatura): indice indiretto di flessibilita’ strutturale.")
6
In un cristallo di proteine, in ogni cella (sito
reticolare) si trova una proteina = somma di densità elettroniche dovute ai singoli atomi
 si trova una proteina = somma di. densità elettroniche dovute ai singoli atomi.")
7
Il risultato sperimentale diretto di un’ analisi cristallografica è una mappa di densità elettronica e non il modello atomico che voi state osservando!!! Se vi sono errori nelle strutture cristallografiche questi sono dovuti all’interpretazione (soggettiva) delle mappe elettroniche da parte del cristallografo!!
 delle mappe elettroniche da parte del cristallografo!!")
10
PARAMETRI X VALUTAZIONE DELL’INFORMAZIONE CONTENUTA NELLA STRUTTURA X-RAY:
Risoluzione (R) della mappa di densità elettronica (tra 3 e 1.5 Å). Strutture con R > 2.5 Å dettagli solo sul backbone, R < 2 Å dettagli su atomi catene laterali R-value e R-free: parametri associati al livello di “discordanza” tra modello atomico e mappa di densita’ elettronica (devono essere R-value < 20% e R-free < 30%). Forniscono, in ultima istanza, indicazioni su quanto differiscono tra loro mappa di densita’ elettronica osservata (sperimentale) e mappa di densita’ elettronica calcolata dal modello della struttura finale.
 della mappa di densità elettronica (tra 3 e 1.5 Å). Strutture con R > 2.5 Å dettagli solo sul backbone, R < 2 Å dettagli su atomi catene laterali. R-value e R-free: parametri associati al livello di discordanza tra modello atomico e mappa di densita’ elettronica (devono essere R-value < 20% e R-free < 30%). Forniscono, in ultima istanza, indicazioni su quanto differiscono tra loro mappa di densita’ elettronica osservata (sperimentale) e mappa di densita’ elettronica calcolata dal modello della struttura finale.")
11
Spettroscopia a risonanza magnetica nucleare (NMR)
Permette di ottenere informazioni sulla struttura di una molecola attraverso l’interazione con una radiazione elettromagnetica. Si basa sullo stesso principio della risonanza magnetica usata in medicina, usa onde radio. I nuclei atomici (elettricamente carichi) ruotano, con una velocità angolare quantizzata, creando un momento magnetico Immersi in un campo magnetico omogeneo esterno i momenti magnetici si allineano (“traballando” a causa del rumore termico). I nuceli vengono irradiati da onde radio (RF), l’effetto è di “disallineare” (tanto da farli “ribaltare”). È possibile rilevare quando i momenti magnetici dei vari nuclei (che continuano a ruotare) si inclinano completamente sul piano perpendicolare rispetto al campo magnetico applicato grazie ad un'antenna che capta le onde radio che questi generano ed è collocata perpendicolarmente al campo magnetico applicato.
 ruotano, con una velocità angolare quantizzata, creando un momento magnetico Immersi in un campo magnetico omogeneo esterno i momenti magnetici si allineano ( traballando a causa del rumore termico). I nuceli vengono irradiati da onde radio (RF), l’effetto è di disallineare (tanto da farli ribaltare ) È possibile rilevare quando i momenti magnetici dei vari nuclei (che continuano a ruotare) si inclinano completamente sul piano perpendicolare rispetto al campo magnetico applicato grazie ad un antenna che capta le onde radio che questi generano ed è collocata perpendicolarmente al campo magnetico applicato.")
12
Spettroscopia a risonanza magnetica nucleare (NMR)
Ogni nucleo mostra le sue caratteristiche perchè ruota a velocità differente a seconda della sua posizione nella molecola e all'ambiente che gli atomi vicini gli fanno sentire e quindi risuona a frequenze radio diverse. Nuclei diversi risuonano a frequenze diverse. Ciò significa innanzitutto che un atomo di carbonio deve essere colpito da un'onda radio con frequenza diversa da quella necessaria ad un atomo di idrogeno per “ribaltarsi” di 90°, ma anche che atomi simili in ambienti diversi, come un atomo di idrogeno legato ad un atomo di ossigeno ed un atomo di idrogeno legato ad un atomo di carbonio si ribaltano a frequenze diverse. Questo è dovuto alla “schermatura” degli elettroni vicini Caratteristiche: studio delle proteine in soluzione (non occorre cristallizzarle) alta risoluzione temporale (millisecondi) informazioni sulle distanze interprotoniche non precise la “proton signature” limita il metodo allanalsi di molecole “piccole” (<30 KD <250 residui) informazioni su strutture di complessi proteici deducibili solo con il ricorso a metodi di modellistica guidati dai dati sperimentali
 alta risoluzione temporale (millisecondi) informazioni sulle distanze interprotoniche non precise. la proton signature limita il metodo allanalsi di molecole piccole (<30 KD <250 residui) informazioni su strutture di complessi proteici deducibili solo con il ricorso a metodi di modellistica guidati dai dati sperimentali")
13
PDB: banca dati di strutture (Protein Data Bank) – ridondante – strutture ottenute sperimentalmente via X-ray o NMR Il file PDB Esempio: Deossiemoglobina umana (1a3n) HEADER OXYGEN TRANSPORT JAN A3N TITLE DEOXY HUMAN HEMOGLOBIN COMPND MOL_ID: 1; COMPND 2 MOLECULE: HEMOGLOBIN; COMPND 3 CHAIN: A, B, C, D; COMPND 4 BIOLOGICAL_UNIT: ALPHA-BETA-ALPHA-BETA TETRAMER SOURCE MOL_ID: 1; SOURCE 2 ORGANISM_SCIENTIFIC: HOMO SAPIENS; SOURCE 3 ORGANISM_COMMON: HUMAN; SOURCE 4 TISSUE: BLOOD; SOURCE 5 CELL: RED CELL KEYWDS OXYGEN TRANSPORT, HEME, RESPIRATORY PROTEIN, ERYTHROCYTE EXPDTA X-RAY DIFFRACTION AUTHOR J.TAME,B.VALLONE REVDAT APR-98 1A3N REMARK REMARK REMARK 2 RESOLUTION ANGSTROMS REMARK 3 […]
 HEADER OXYGEN TRANSPORT 22-JAN-98 1A3N TITLE DEOXY HUMAN HEMOGLOBIN COMPND MOL_ID: 1; COMPND 2 MOLECULE: HEMOGLOBIN; COMPND 3 CHAIN: A, B, C, D; COMPND 4 BIOLOGICAL_UNIT: ALPHA-BETA-ALPHA-BETA TETRAMER SOURCE MOL_ID: 1; SOURCE 2 ORGANISM_SCIENTIFIC: HOMO SAPIENS; SOURCE 3 ORGANISM_COMMON: HUMAN; SOURCE 4 TISSUE: BLOOD; SOURCE 5 CELL: RED CELL KEYWDS OXYGEN TRANSPORT, HEME, RESPIRATORY PROTEIN, ERYTHROCYTE EXPDTA X-RAY DIFFRACTION AUTHOR J.TAME,B.VALLONE REVDAT 1 29-APR-98 1A3N 0 REMARK 1 REMARK 2 REMARK 2 RESOLUTION. 1.8 ANGSTROMS. REMARK 3. […]")
14
tipo di atomo tipo di amminoacido coordinate X Y Z …
ATOM N VAL A N ATOM CA VAL A C ATOM C VAL A C ATOM O VAL A O ATOM CB VAL A C ATOM CG1 VAL A C ATOM CG2 VAL A C ATOM N LEU A N ATOM CA LEU A C ATOM C LEU A C ATOM O LEU A O ATOM CB LEU A C ATOM CG LEU A C ATOM CD1 LEU A C ATOM CD2 LEU A C ATOM N SER A N ATOM CA SER A C ATOM C SER A C ATOM O SER A O ATOM CB SER A C ATOM OG SER A O …

15
CONFRONTO STRUTTURE 3D DI PROTEINE – ALLINEAMENTO STRUTTURALE
Come nel confronto di sequenze e’ necessario allinearle, nel confronto di strutture 3D e’ necessario sovrapporle come corpi rigidi scegliendo una regola di corrispondenza tra coppie di atomi o di residui nelle due strutture. La prima difficoltà consiste nel fatto che le due proteine molto spesso non hanno lo stesso numero di residui. Per la sovrapposizione si possono utilizzare le catene dei carboni alfa appartenenti agli elementi di struttura secondaria perche’ in genere le inserzioni e delezioni si accumulano nei loop che possono semplicemente venire esclusi dalla sovrapposizione. I metodi di confronto 3D utilizzano l’ allineamento delle sequenze per decidere la regola di corrispondenza alla base della sovrapposizione strutturale

16
Un allineamento strutturale può essere valutato in base alla deviazione quadratica media (root mean square deviation o r.m.s.d.), al numero di atomi che sono stati accoppiati nella sovrapposizione e alla valutazione della similarità dei residui sovrapposti. L’r.m.s.d. o r.m.s. di una sovrapposizione tridimensionale è la distanza media tra gli atomi di tutte le coppie che hanno partecipato all’allineamento strutturale, per cui tanto più bassa è l’r.m.s. tanto migliore sarà l’allineamento strutturale calcolato D = distanza tra coppie di atomi appaiati N = numero di coppie considerate

17
RMSD Root-mean-square deviation
rai e rbi sono le posizioni dell´ atomo i nelle strutture a e b, n è il numero di atomi nelle strutture. r.m.s.d. di una sovrapposizione 3D è la distanza media tra gli atomi di tutte le coppie che hanno partecipato all’allineamento strutturale, per cui tanto più bassa è l’r.m.s. tanto migliore sarà l’allineamento strutturale calcolato Root-mean-square deviation Deviazione quadratica media Serve per paragonare strutture identiche, eccetto rotazioni e traslazioni

18
EVOLUZIONE DELLE PROTEINE
RELAZIONE ESISTENTE TRA SIMILARITA’ di sequenza e struttura in omologhi Studio di un campione di strutture note di proteine omologhe (Chothia, Lesk, 1982) Similarità strutturale misurata in termini di RMSD CALCOLO RMS Confronto di due strutture mediante SOVRAPPOSIZIONE Determinazione di aa che si corrispondono nelle due strutture Identificazione degli atomi che si vogliono confrontare (Cα o backbone) RMSD= n = numero residui d = distanza tra atomi corrispondenti
 Similarità strutturale misurata in termini di RMSD. CALCOLO RMS. Confronto di due strutture mediante SOVRAPPOSIZIONE. Determinazione di aa che si corrispondono nelle due strutture. Identificazione degli atomi che si vogliono confrontare (Cα o backbone) RMSD= n = numero residui. d = distanza tra atomi corrispondenti.")
19
valutazione dell’allineamento strutturale
un altro criterio di valutazione di un allineamento strutturale è rappresentato dal numero di atomi o di residui che sono stati accoppiati si cerca di massimizzare il numero di atomi accoppiati e di minimizzare la corrispondente r.m.s. a parità di numero di residui accoppiati, il migliore allineamento strutturale sarà quello con minore r.m.s. a parità di r.m.s. verrà considerato migliore l’allineamento strutturale operato con un maggior numero di atomi accoppiati oltre a questi due valori tipici delle sovrapposizioni tridimensionali, si può anche considerare il punteggio di similarità dei residui accoppiati

20
DIVERGENZA DI SEQUENZA E STRUTTURA
Se si valuta la relazione esistente tra divergenza strutturale (misurata in termini di RMSD) e divergenza di sequenza si osserva che esse si corrispondono in MODO ESPONENZIALE (valutato dal confronto di strutture note) 30% id.seq RMSD (A) %identity (RMSD< 2 Å) possono avere seq molto differenti (degenerazione del codice strutturale-struttura più conservata della seq)
 e divergenza di sequenza si osserva che esse si corrispondono in MODO ESPONENZIALE (valutato dal confronto di strutture note) 30% id.seq. RMSD (A) %identity. (RMSD< 2 Å) possono avere seq molto differenti. (degenerazione del codice strutturale-struttura più conservata della seq)")
21
SOGLIA di significatività per la SIMILARITA’ STRUTTURALE
.confronto a coppie di struttura e seq di proteine 3D note, quantificato usando come parametri: lunghezza allineamento, % id seq, sim.struttura a confronto in un grafico: lunghezza allin vs id seq (e in terza dimensione sim.strutturale) Sim. Strut.parametrizzata come uguale/diverso (pallino/quadrato): 2 str. Uguali se > 70% della struttura sec è in comune (rmsd bassa). al di sotto della soglia rumore di fondo elevato 25% (x allin 80 aa) al di sopra del quale chiara similarità strutturale – soglia lunghezza dipendente possibile che proteine con identità < 25% abbiano strutture simili ma anche che non siano correlate strutturalmente (twilight zone) 100 Sec str identity < 70% %id seq Sec str identity > 70% 30 Length alignment 150
 Sim. Strut.parametrizzata come uguale/diverso (pallino/quadrato): 2 str. Uguali se > 70% della struttura sec è in comune (rmsd bassa). al di sotto della soglia rumore di fondo elevato. 25% (x allin 80 aa) al di sopra del quale chiara similarità strutturale – soglia lunghezza dipendente. possibile che proteine con identità < 25% abbiano strutture simili ma anche che non siano correlate strutturalmente (twilight zone) 100. Sec str identity < 70% %id seq. Sec str identity > 70% 30. Length alignment")
22
Dalla sequenza al modello
Raw model Loop modeling Side chain placement Refinement

23
HOMOLOGY MODELLING OMOLOGO 3D (PDB) ALLINEAMENTO RICERCA DEL TEMPLATO
Blast-FastA CRITERI IDENTITA’/SIMILARITA’ CONOSCENZA FUNZ.-STR.-BIOCHIM.

24
ALLINEAMENTO GUIDA LA COSTRUZIONE DEL MODELLO
CORRISPONDENZA aa target aa templato ricerca ALLINEAMENTO OTTIMALE CORRISPONDENZA DI aa FUNZ. IMPORTANTI CORRISPONDENZA DELLA STRUTTURA SECONDARIA TRA TEMPLATO E QUERY VALUTAZIONE DEI GAP loop USO TEMPLATI MULTIPLI loc.similarità

25
ALLINEAMENTO 1. Generazione di allineamenti a coppie diversi con diversi programmi (nel caso di un solo templato) 2. Allineamenti multipli di omologhi per avere informazioni maggiori 3. Ricerca di informazioni biologico-biochimiche sugli aa conservati 4. Predizione di struttura secondaria per il target 5. Correzione dell’allineamento sulla base delle informazioni ai punti e 4.

26
CREAZIONE DEL MODELLO ______________ ______________ x-ray SCRs
identificazione SCR (structural conserved regions) SCR scaffold del modello Raw model Loop modeling Side chain placement Refinement ______________ ______________ x-ray SCRs No SCRs (loops ?)
 SCR scaffold del modello. Raw model. Loop modeling. Side chain placement. Refinement. ______________. ______________ x-ray. SCRs. No SCRs (loops )")
27
Costruzione del pre-modello
La struttura del templato viene utilizzata come “stampo“ per costruire il modello seguendo l‘allineamento. Le coordinate 3D dei residui strutturalmente conservati si possono copiare direttamente. Le regioni variabili della struttura (generalmente loop) non si possono copiare. flexible conserved
 non si possono copiare. flexible. conserved.")
28
Catene laterali Problema: Applicando le coordinate del templato sulla sequenza del target cambiano tipo, dimensione e posizione delle catene laterali. L‘RMSD cambia relativamente poco, però possono cambiare le conformazioni di residui importanti (p.es. del sito attivo) Dove possibile è meglio mantenere le conformazioni delle catene laterali del templato. Esistono metodi standard per risolvere questo problema. Raw model Loop modeling Side chain placement Refinement
 Dove possibile è meglio mantenere le conformazioni delle catene laterali del templato. Esistono metodi standard per risolvere questo problema. Raw model. Loop modeling. Side chain placement. Refinement.")
29
PREDIZIONE DELLE CATENE LATERALI
AUSILIO DI LIBRERIE DI ROTAMERI Contengono i possibili conformeri delle catene laterali a fronte di specifiche conformazioni del backbone OTTIMIZZAZIONE ENERGETICA DELLE STRUTTURA rimozione di clash

30
Loop modeling Al pre-modello possono mancare interi frammenti di catena principale non conservati nella famiglia proteica Inserzioni Delezioni Descrizione del problema: Si cerca un fold che colleghi il frammento N-terminale (pre-loop) con quello C-terminale (post-loop) tramite k residui (f,y) sono gli unici parametri liberi Raw model Loop modeling Side chain placement Refinement loop post-loop pre-loop
 con quello C-terminale (post-loop) tramite k residui. (f,y) sono gli unici parametri liberi. Raw model. Loop modeling. Side chain placement. Refinement. loop. post-loop. pre-loop.")
31
PREDIZIONE DEI LOOP REGIONI VARIABILI – pressione selettiva
DUE STRATEGIE PREDITTIVE: criterio geometrico testando diverse conformazioni ricerca in PDB di frammenti simili nelle proteine a struttura nota INFLUENZA DELL’INTORNO REFINEMENT CRITICO OTTIMIZZAZIONE MEC. E DIN. MOL. CRITICITA’ nella predizione quando i LOOP rivestono ruolo funzionale o di interazione.

32
5. Ottimizzazione del modello
Regolarizzazione di legami, angoli e torsioni Eliminazioni di clash strutturali Minimizzazione energetica









![PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]](/1/540371/big_thumb.jpg "PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]>")

 tra la radiazione.>")








>")

 LABORATORIO LASER (prof. R. Calabrese, dott. L. Tomassetti) LABORATORIO DI.>")
, CERN, JLAB) 1) Trasparenza di colore 2) Adronizzazione Perché>")