Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Fisica Computazionale applicata alle Macromolecole
Metodi di allineamento Pier Luigi Martelli Università di Bologna

2
Ricordiamo... La struttura di una proteina in ambiente fisiologico dipende solo dalla sua sequenza amminoacidica Esperimento di Anfinsen Differenti sequenze amminoacidiche assumono in ambiente fisiologico lo stesso fold Banche dati CATH e SCOP: Organizzazione gerarchica La funzione dipende dalla struttura, sebben questa relazione non sia rigorosa La funzione dipende da tutta la struttura, non solo dal fold

3
Problema Determinare il fold a partire dalla sequenza amminoacidica Allineare le sequenze in modo da riprodurre l’allineamento generato dalla sovrapposizione delle loro due strutture Strategia Esaminare come le sequenze hanno avuto origine Perché nei vari organismi esistono sequenze differenti per una unica funzione?

4
Evoluzione Molecolare e omologia
Evoluzione: Mutabilità e Selezione Naturale Le sequenze degli organismi attuali hanno avuto origine dall’evoluzione di sequenze ancestrali Le sequenze genomiche cambiano continuamente in modo casuale L’ambiente seleziona gli individui in base al loro fenotipo Se il prodotto del gene modificato non è funzionale (perde struttura o funzione) l’individuo muore e la modifica non si trasmette NB. Le mutazioni sono casuali? Almeno la loro velocità, non sempre: SOS polimerasi di Radman
 l’individuo muore e la modifica non si trasmette. NB. Le mutazioni sono casuali Almeno la loro velocità, non sempre: SOS polimerasi di Radman.")
5
Evoluzione Molecolare e omologia
Due sequenze sono dette omologhe se hanno un ancestore comune Ortologhe in due specie differenti Paraloghe all’interno della stessa specie (duplicazione genica) Similarità Due sequenze sono dette simili se condividono buona parte della sequenza (molti amminoacidi uguali o simili): concetto NON evolutivo, ma di confronto tra sequenze
 Similarità. Due sequenze sono dette simili se condividono buona parte della sequenza (molti amminoacidi uguali o simili): concetto NON evolutivo, ma di confronto tra sequenze.")
6
Omologia e Similarità Sequenze omologhe sono sempre simili? Dipende dal grado di divergenza Sequenze simili sono sempre omologhe? Sequenze differenti possono essere evolute convergentemente verso sequenze simili (es., non su sequenze, ali di uccelli e ali di pipistrello sono evoluzioni convergenti, a partire da da rettili e da mammiferi) Di principio similarità e omologia non coincidono esattamente. Tuttavia se due sequenze sono molto simili sono probabilmente omologhe. Per ora misuriamo la similarità in termini di identità di sequenza
 Di principio similarità e omologia non coincidono esattamente. Tuttavia se due sequenze sono molto simili sono probabilmente omologhe. Per ora misuriamo la similarità in termini di identità di sequenza.")
7
Identità di sequenza e identità strutturale
Quando la similarità di sequenza implica similiarità strutturale?

8
Identità di sequenza e identità strutturale
Fino a quanto due sequenze simili danno strutture uguali? 2.5 0.0 1.0 0.2 0.6 0.8 0.4 100 50 Percent identical residues in core Fraction of residues in core with RMSD < 0.1 nm 2.0 1.5 Rmsd of backbone atoms in core 1.0 0.5 0.0 100 50 Percent identical residues in core 2 proteine sono sovrapposte e si esamina la percentuale di identità nel nucleo sovrapposto Proteine con identità maggiore del 60% hanno il 90% dei residui sovrapposti a meno di 0.1 nm Chothia, C. & Lesk, A. M. (1986). The relation between the divergence of sequence and structure in proteins. EMBO J. 5,
. The relation between the divergence of sequence and structure in proteins. EMBO J. 5,")
9
Identità di sequenza e identità strutturale
2 4 6 8 1 . i d e n t i t y Identità di sequenza implica identità strutturale Identità di sequenza (%) Identità di sequenza NON implica identità strutturale 5 1 2 Numero di residui allineati Rost B (1999). The twilight zone of protein alignments. Protein Engineering 12,
 Identità. di sequenza. NON implica identità strutturale Numero di residui allineati. Rost B (1999). The twilight zone of protein alignments. Protein Engineering 12,")
10
Identità di sequenza e identità strutturale
Quindi due sequenze più lunghe di 100 residui, che condividano il 30 % dei residui, hanno struttura simile Per sequenze più corte la percentuale di identità deve essere più alta Questo NON implica che sequenze con identità minore abbiano strutture differenti Esempio: Mioglobina di capodoglio e emoglobina batterica: RMSD = 0.19 nm, Identità: 14% .

11
Identità di sequenza e identità strutturale
Per sequenze più lunghe di 100 residui Midnight zone: contiene la maggior parte delle proteine strutturalmente simili Twilight zone: alto numero di falsi positivi (sequenza simile struttura diversa) Safe zone: nessun falso positivo tutte le sequenze simili hanno la stessa struttura 20% 30% Percentuale di identità
 Safe zone: nessun falso positivo. tutte le sequenze. simili hanno la. stessa struttura. 20% 30% Percentuale di identità.")
12
Allineamento di sequenze
Problema: date due sequenze, confrontarle in modo da rilevare la loro similarità Definire una distanza tra le sequenze Cercare un algoritmo per trovare l’allineamento a minima distanza Studiare metodi per validare la significativicità dell’allineamento

13
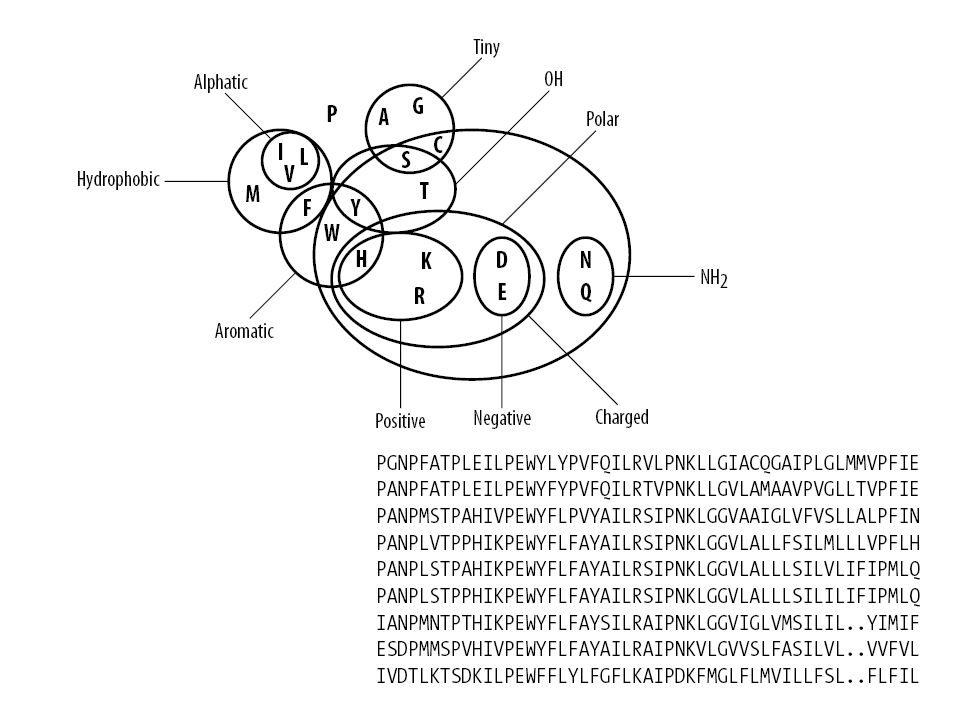
Distanza tra sequenze Quali eventi consideriamo? Mutazione Va definito un punteggio per la sostituzione dell’amminoacido i con l’amminoacido j Matrici di sostituzione s(i,j) A: ALASVLIRLITRLYP B: ASAVHLNRLITRLYP La matrice di sostituzione riflette se una mutazione è mediamente compatibile col folding e col mantenimento della funzione
 A: ALASVLIRLITRLYP. B: ASAVHLNRLITRLYP. La matrice di sostituzione riflette se una mutazione è mediamente compatibile col folding e col mantenimento della funzione.")
15
Derivazione degli score da allineamenti di sequenze omologhe
Vogliamo misurare la probabilità di mutazione di ogni tipo di amminoacido in un insieme di sequenze omologhe Date (molte) coppie di sequenze correlate, misuriamo la frequenza della sostituzione iA->jB o iB->jA (indipendente dalla direzione): Pij Es: A: ALASVLIRAILRLYP B: ALAVLLNRLILRALP P(A,A)= N(AA,AB)/N = 2/15 P(A,L)= P(L,A)= [N(LA,AB)+N(AA,LB)]/N = 2/15
 coppie di sequenze correlate, misuriamo la frequenza della sostituzione iA->jB o iB->jA (indipendente dalla direzione): Pij. Es: A: ALASVLIRAILRLYP. B: ALAVLLNRLILRALP. P(A,A)= N(AA,AB)/N = 2/15. P(A,L)= P(L,A)= [N(LA,AB)+N(AA,LB)]/N = 2/15.")
16
La sostituzione è significativa?
Qual è la probabilità che la sostituzione i->j sia casuale (e quindi non significativa)? Es: 1° insieme di sequenze omologhe A: ALASVLIRAILRLYP B: ALAVLLNRLILRALP 2° insieme di sequenze omologhe A: LLLLAALLLALLALL B: LALLAALLAALLALL P(A,L)= 2/15 in entrambi i casi. Sono ugualmente significativi? La probabilità che questa sostituzione sia casuale dipende dalle frequenze di occorrenza dei singoli amminoacidi Pi e Pj
 Es: 1° insieme di sequenze omologhe. A: ALASVLIRAILRLYP. B: ALAVLLNRLILRALP. 2° insieme di sequenze omologhe. A: LLLLAALLLALLALL. B: LALLAALLAALLALL. P(A,L)= 2/15 in entrambi i casi. Sono ugualmente significativi La probabilità che questa sostituzione sia casuale dipende dalle frequenze di occorrenza dei singoli amminoacidi Pi e Pj.")
17
Confronto con l’ipotesi di indipendenza
Sostituzione iA -> jB casuale significa che i 2 eventi: E1 = (i in A) e E2 = (j in B) sono INDIPENDENTI Per determinare il grado di “non casualità” della sostituzione bisogna confrontare Pij con il prodotto PiPj Es: 1° insieme di sequenze omologhe A: ALASVLIRAILRLYP B: ALAVLLNRLILRALP P(A)= 6/30, P(L) =10/30 P(A,L) = 2/15 > 1/15 = P(A)P(L): sostituzione FAVORITA 2° insieme di sequenze omologhe A: LLLLAALLLALLALL B: LALLAALLAALLALL P(A)= 10/30, P(L) =20/30 P(A,L) = 2/15 < 2/9 = P(A)P(L): sostituzione SFAVORITA
 e E2 = (j in B) sono INDIPENDENTI. Per determinare il grado di non casualità della sostituzione bisogna confrontare Pij con il prodotto PiPj. Es: 1° insieme di sequenze omologhe. A: ALASVLIRAILRLYP. B: ALAVLLNRLILRALP. P(A)= 6/30, P(L) =10/30. P(A,L) = 2/15 > 1/15 = P(A)P(L): sostituzione FAVORITA. 2° insieme di sequenze omologhe. A: LLLLAALLLALLALL. B: LALLAALLAALLALL. P(A)= 10/30, P(L) =20/30. P(A,L) = 2/15 < 2/9 = P(A)P(L): sostituzione SFAVORITA.")
18
Score di sostituzione Il rapporto rij = Pij/PiPj determina se la sostituzione i -> j è più o meno frequente di quanto ci si aspetterebbe casualmente. Dato un allineamento tra due sequenze: A: SLDPIKHTYRALMNVDSLRTFPIL B: SFGIKKHTKLAKLPVDTIKSWPIL la probabilità di sostituzione A->B sarà data dal prodotto degli rij : rSS rLF rDG rPI rIK … (indipendenza delle posizioni) SCORE di SOSTITUZIONE: s(i,j) =int[K log(Pij/PiPj)] Il logaritmo rende la quantità additiva sulla sequenza Minima distanza = Massimo score (s)
 SCORE di SOSTITUZIONE: s(i,j) =int[K log(Pij/PiPj)] Il logaritmo rende la quantità additiva sulla sequenza. Minima distanza = Massimo score (s)")
19
ESERCIZIO Calcolare la matrice di sostituzione a partire dalle seguenti sequenze allineate ACAGGTGGACCT ACTGGTCGACTT CTATATGG CCGGATCG

20
Matrici di sostituzione: PAM
In base a questo concetto, differenti matrici possono essere derivate. La differenza fondamentale sta nell’insieme di allineamenti considerati per costruire le matrici. PAMx: (Point Accepted Mutation). Numero di eventi mutazionali pari a x%. Si costruisce la matrice: A1ij = P(j|i) = N(i,j)/N(i) per sequenze con 1% di mutazioni. PAM 1 = Log(A1ij /Pi)
. Numero di eventi mutazionali pari a x%. Si costruisce la matrice: A1ij = P(j|i) = N(i,j)/N(i) per sequenze con 1% di mutazioni. PAM 1 = Log(A1ij /Pi)")
21
Matrici di sostituzione: PAM
Per derivare gli score relativi a sequenze in cui siano avvenuti n eventi mutazionali ogni 100 residui: Anij=(A1ij)n Es: n=2 P(i|j) = Pl P(i|l) P(l|j) NOTA BENE: n % eventi mutazionali: numero di mutazioni, NON di residui mutati. Possono essere rimutati posizioni già mutate. 100 eventi mutazionali indipendenti ogni 100 residui lasciano alcune posizioni invariate PAM n = Log(Anij /Pi)
n. Es: n=2 P(i|j) = Pl P(i|l) P(l|j) NOTA BENE: n % eventi mutazionali: numero di mutazioni, NON di residui mutati. Possono essere rimutati posizioni già mutate. 100 eventi mutazionali indipendenti ogni 100 residui lasciano alcune posizioni invariate. PAM n = Log(Anij /Pi)")
22
Relazione tra PAM e identità tra due sequenze
Il numero di eventi mutazionali (PAM) è differente dal numero di residui differenti tra due sequenze, quando le mutazioni si accumulano.
 è differente dal numero di residui differenti tra due sequenze, quando le mutazioni si accumulano.")
23
PAM10 Matrice molto stringente: nessun valore positivo fuori diagonale

24
PAM160 Iniziano valori positivi fuori diagonale: residui con valori di sostituzione positivi sono detti SIMILI

25
PAM250 Molto usata

26
PAM500

27
Matrici di sostituzione
Le matrici PAM ricavano ipotesi sulle mutazioni in sequenze lontane a partire dalle mutazioni osservate in sequenze molto simili. Ipotesi molto stretta. BLOSUMx: Famiglia di matrici ricavate direttamente da allineamenti di sequenze con identità maggiore al x%. Per sequenze molto relate vanno usate PAM basse o BLOSUM alte. Per sequenze lontane, viceversa.

28
BLOSUM62 Molto usata

29
BLOSUM90

30
BLOSUM30

31
Distanza tra sequenze Quali eventi consideriamo? Mutazione Delezione e Inserzione Alcuni amminoacidi possono essere stati deleti o inseriti nel corso dell’evoluzione A: ALASVLIRLIT--YP B: ASAVHL---ITRLYP Il punteggio (negativo) di un gap dipende solo dal numero di posizioni s(n) = -nd lineare s(n) = -d - (n-1)e affine (d: apertura, e: estensione) N.B. Tutti i punteggi sono indipendenti dalla posizione lungo la sequenza
 di un gap dipende solo dal numero di posizioni. s(n) = -nd lineare. s(n) = -d - (n-1)e affine (d: apertura, e: estensione) N.B. Tutti i punteggi sono indipendenti dalla posizione lungo la sequenza.")
32
Allineamento tra sequenze
Date due sequenze, qual è l’allineamento a punteggio massimo? Soluzione naïf: provare tutti gli allineamenti possibili e scegliere quello a punteggio maggiore! Per ogni allineamento, possiamo infatti calcolare il punteggio tramite la formula

33
Quanti sono i possibili allineamenti di due sequenze?
Scrivere TUTTI i possibili allineamenti senza gap interni delle sequenze: A: tca B: ga Scrivere TUTTI i possibili allineamenti con gap delle medesime sequenze Scrivere i punteggi di allineamento per ognuno degli allineamenti secondo la seguente matrice con penalità di gap LINEARE (d=2)
")
34
Quanti sono i possibili allineamenti di due sequenze?
Caso senza Gap interni --tca -tca tca tca tca- tca-- ga--- ga-- ga- -ga --ga ---ga Date due sequenze di lunghezza m e n, il numero dei possibili scorrimenti differenti è m +n Uguale al primo

35
Quanti sono i possibili allineamenti di due sequenze?
Caso con gap interni --tca -tca -tca -tca t-ca ga--- ga-- g-a- g--a ga-- gatca gtaca gtcaa gtcaa tgaca tca tca tc-a tca tca- ga- g-a -ga- -ga --ga tgcaa tgcaa tcgaa tcgaa tcaga I possibili allineamenti sono uguali ai possibili modi di intercalare le due sequenze, mantenendo l’ordine Date due sequenze di lunghezze n e m, i possibili allineamenti sono (m+n)!/n!m! Per n=m=80 ho 9•1042 possibili allineamenti !!!!!!!
!/n!m! Per n=m=80 ho 9•1042 possibili allineamenti !!!!!!!")
36
Algoritmi di programmazione dinamica: idea base
Il calcolo per intero di tutti gli allineamenti è sovrabbondante ALSKLASPALSAKDLDSPALS ALSKIADSLAPIKDLSPASLT ALSKLASPALSAKDLDSPAL-S ALSKIADSLAPIKDLSPASLT- I due allineamenti sono per la maggior parte uguali. Lo score è additivo lungo l’allineamento. Col metodo naïf la prima parte dell’allineamento viene ricalcolata! Si possono memorizzare i punteggi degli allineamenti parziali

37
Algoritmi di programmazione dinamica: idea base
Costruire l’allineamento per passi Date le due sequenze ALSKLASPALSAKDLDSPALS, ALSKIADSLAPIKDLSPASLT il miglior allineamento tra le sottostringhe ALSKLASPA ALSKIAD deriva dai migliori allineamenti ALSKLASP A ALSKLASP A ALSKLASPA - ALSKIA D ALSKIAD - ALSKIA D Ed è il migliore dei tre! (additività dei punteggi sulle posizioni) + + +
")
38
Algoritmo di Needleman e Wunsch
Allineamento globale di sequenze, gap a penalità lineare Date due sequenze A e B, di lunghezze a e b, definiamo la matrice F(i,j): punteggio del miglior allineamento tra le sottosequenze: A1A2A3…….Ai e B1B2B3…….Bj. Inizializzazione F(0,0) = 0 F(i-1,j-1) + s(Ai,Bj) Iterazione F(i,j) = Max F(i-1,j) - d F(i,j-1) - d ALSKLASP A ALSKLASP A ALSKLASPA - ALSKIA D ALSKIAD - ALSKIA D F(i-1,j-1) F(i-1,j) F(i,j-1) Iterazione F(a,b) è il punteggio del miglior allineamento + + +
: punteggio del miglior allineamento tra le sottosequenze: A1A2A3…….Ai e B1B2B3…….Bj. Inizializzazione F(0,0) = 0. F(i-1,j-1) + s(Ai,Bj) Iterazione F(i,j) = Max F(i-1,j) - d. F(i,j-1) - d. ALSKLASP A ALSKLASP A ALSKLASPA - ALSKIA D ALSKIAD - ALSKIA D. F(i-1,j-1) F(i-1,j) F(i,j-1) Iterazione F(a,b) è il punteggio del miglior allineamento")
39
Algoritmo di Needleman e Wunsch
Allineare le sequenze ACTGG e ACCA

40
Algoritmo di Needleman e Wunsch

41
Algoritmo di Needleman e Wunsch
+2 -2 MAX

42
Algoritmo di Needleman e Wunsch

43
Algoritmo di Needleman e Wunsch
Punteggio del miglior allineamento Gap in sequenza 2 Gap in sequenza 1 Match 0 A C T G G 0 A C C A - 0 A C T G G 0 A C C - A

44
Complessità computazionale
Numero di operazioni necessario per ottenere un risultato seguendo un algoritmo Algoritmo naïf Date due sequenze di lunghezza n dobbiamo calcolare (2n)!/(n !)2 punteggi di allineamento. Ognuno richiede dalle n alle 2n operazioni. Poiché n ! n n (2 n)1/2 e-n Complessità O(22n n 1/2) Algoritmo Needleman-Wunsch Vanno calcolati (n +1)2 valori della matrice. Ognuno richiede 4 operazioni: Complessità (n 2)
!/(n !)2 punteggi di allineamento. Ognuno richiede dalle n alle 2n operazioni. Poiché n ! n n (2 n)1/2 e-n. Complessità O(22n n 1/2) Algoritmo Needleman-Wunsch. Vanno calcolati (n +1)2 valori della matrice. Ognuno richiede 4 operazioni: Complessità (n 2)")
45
Allineamenti locali Quelli visti fino ad ora sono allineamenti GLOBALI (tutta la sequenza) Se volessimo cercare solo le zone di miglior sovrapposizione (domini comuni, elementi funzionali conservati…)? La strategia è la medesima. Solo si eliminano i punteggi negativi (meglio riiniziare un allineamento che portarlo a punteggi negativi)
 La strategia è la medesima. Solo si eliminano i punteggi negativi (meglio riiniziare un allineamento che portarlo a punteggi negativi)")
46
Algoritmo di Smith e Waterman
Allineamento locale di sequenze, gap a penalità lineare Date due sequenze A e B, di lunghezze a e b, definiamo la matrice F(i,j): punteggio del miglior allineamento locale tra le sottosequenze: A1A2A3…….Ai e B1B2B3…….Bj. Inizializzazione F(0,0) =0 F(i-1,j-1) + s(Ai,Bj) Iterazione F(i,j) = Max F(i-1,j) - d F(i,j-1) - d Iterazione Il punteggio F(i,j) massimo sulla matrice dà il miglior allineamento locale
: punteggio del miglior allineamento locale tra le sottosequenze: A1A2A3…….Ai e B1B2B3…….Bj. Inizializzazione F(0,0) =0. F(i-1,j-1) + s(Ai,Bj) Iterazione F(i,j) = Max F(i-1,j) - d. F(i,j-1) - d. Iterazione Il punteggio F(i,j) massimo sulla matrice dà il miglior allineamento locale.")
47
Algoritmo di Smith e Waterman

48
Algoritmo di Smith e Waterman
Gap in sequenza 2 Gap in sequenza 1 Match 0 A C T 0 A T T

49
Significatività di un allineamento
Dato un allineamento (globale o locale) che abbia ottenuto un punteggio S, come valutare se è significativo? Come sono distribuiti i punteggi di allineamenti di sequenze casuali? Con 100,000 allineamenti di sequenze scorrelate e randomizzate: Occorrenza Gli allinementi significativi sono qua! Score
 che abbia ottenuto un punteggio S, come valutare se è significativo Come sono distribuiti i punteggi di allineamenti di sequenze casuali Con 100,000 allineamenti di sequenze scorrelate e randomizzate: Occorrenza. Gli allinementi significativi sono qua! Score.")
50
Z-score Z=(S-<S>)/s S=Punteggio di allineamento <S>=Media dei punteggi di allineamento su un insieme random s=Deviazione dei punteggi di allineamento su un insieme random Accuratezza dell’allineamento Z<3 non significativo 3<Z<6 putativamente significativo 6<Z<10 possibilmente significativo Z>10 significativo

51
Quanto è affidabile lo Z-score?
Lo Z-score di questo allineamento locale è 7.5 su 54 residui L’identità è 25.9%. Le sequenze sono completamente differenti in struttura secondaria Citrate synthase (2cts) vs transthyritin (2paba)
 vs transthyritin (2paba)")
52
E-value Numero atteso di allineamenti random con punteggio maggiore o uguale a un punteggio dato (s) E’ reso possibile dal calcoli statistici E=Kmn e-ls m, n: lunghezze delle due sequenze K, l: Costanti di “scaling” Il numero di allineamenti random a punteggio maggiore di s cresce col crescere delle lunghezze delle sequenze (o dei data base con cui confrontiamo una sequenza) e cala esponenziamente al crecere di s
 e cala esponenziamente al crecere di s.")
53
E-value Accuratezza dell’allineamento La significatività dell’E-value dipende dalla lunghezza della banca dati considerata. Per un numero di sequenze pari a quello di SwissProt E> non significativo 10-1 > E > putativamente significativo 10 -3 > E > possibilmente significativo E < altamente significativo

54
Programmi di allineamento a coppie: LALIGN
1BVD.seq 1MWD.seq

55
Allineamento globale: Sequenze simili
1BVD: mioglobina di capodoglio 1MWD: mioglobina di maiale

56
L’allineamento corrisponde all’allineamento strutturale?

57
L’allineamento corrisponde all’allineamento strutturale?

58
Allineamento locale: Sequenze simili

59
Allineamento globale: Sequenze differenti
1BVD: mioglobina di capodoglio 1VHB: emoglobina batterica 1BVD.seq 1VHB.seq

60
L’allineamento corrisponde all’allineamento strutturale?

61
L’allineamento corrisponde all’allineamento strutturale?

62
Allineamento locale: Sequenze differenti

63
Allineamenti Multipli
Il confronto di più sequenze omologhe, può mettere in luce caratteristiche che non emergono da un allineamento a coppie. Allineare molte sequenze Date M sequenze Ai , si può definire come allineamento multiplo (globale) ottimo quello che massimizza lo score S=Si<j S(Ai,Aj) Metodi esatti Esistono metodi di programmazione dinamica multidimensionale che trovano la soluzione ottima. Sono troppo lenti Allineamento progressivo
 ottimo quello che massimizza lo score. S=Si<j S(Ai,Aj) Metodi esatti. Esistono metodi di programmazione dinamica multidimensionale che trovano la soluzione ottima. Sono troppo lenti. Allineamento progressivo.")
64
Allineamenti Multipli Progressivi
Allineamento a coppie e raggruppamento A A C B B C D D Allineamento esatto delle sequenze più simili secondo l’albero Allineamento progressivo dei profili derivati dagli allineamenti effettuati

65
CLUSTALW

66
Allineamento di un insieme di mioglobine (più l’emoglobina)
Sequenze di Mioglobina Allineamento con CLUSTALW

67
L’allineamento multiplo migliora l’allineamento tra due sequenze
1BVD VLSEGEWQLVLHVWAKVEAD---VAGHGQDILIRLFKSHPETLEKFDRFKH 48 1VHB MLDQQTINIIKATVPVLKEHG---VTITTTFYKNLFAKHPEVRPLFDMGR- 47 1BVD LKTEAEMKASEDLKKHGVTVLTALGAILKKKGHHEAELKPLAQSHATKHKIP-----IKY 103 1VHB QESLEQPKALAMTVLAAAQNIENLPAILPAVKKIAVKHCQAG---VAAAH 94 1BVD LEFISEAIIHVLHSRHPGDFGADAQGAMNKALELFRKDIAAKYKELGYQG-- 153 1VHB YPIVGQELLGAIKEVLGDAATDDILDAWGKAYGVIADVFIQVEADLYAQAVE 146 Allineamento estratto dall’allineamento multiplo effettuato da CLUSTALW

68
Ricerca di similarità in Banche Dati
Data una sequenza, cercare se esistono sequenze simili in una banca dati Di principio si potrebbero fare allineamenti tra la sequenza target e TUTTE le sequenze Le sequenze da allineare sono troppe, e il processo non è fattibile in tempi brevi nemmeno usando l’algoritmo di NW Si utilizzano algoritmi euristici, che non assicurano il raggiungimento dell’allineamento ottimo FASTA BLAST

69
FASTA Data una sequenza (Query), viene divisa in “parole” lunghe k-tup (generalmente k-tup = 2 per proteine, 6 per DNA) ADKLPTLPLRLDPTNMVFGHLRI Parole (indicizzate per posizione): AD, DK, KL, LP, PT, TL, LP, PR, RL, …,…, …. Lo stesso elenco di parole indicizzato è compilato per ogni sequenza (Subject) del data base in cui si cercano sequenze. E’ molto rapida la ricerca di parole uguali tra Query e Subject. La differenza degli indici determina la diagonale
: AD, DK, KL, LP, PT, TL, LP, PR, RL, …,…, …. Lo stesso elenco di parole indicizzato è compilato per ogni sequenza (Subject) del data base in cui si cercano sequenze. E’ molto rapida la ricerca di parole uguali tra Query e Subject. La differenza degli indici determina la diagonale.")
70
FASTA Query Subject Identificazione delle identità di “parole”: identità consecutive danno origine a diagonali più lunghe

71
FASTA Query Subject I punteggi delle regioni più lunghe sono valutati con una matrice di score (PAM o BLOSUM)
 .")
72
FASTA Query Subject Vengono cercate regioni ad alta similarità su diagonali vicine

73
FASTA Query Subject Si procede ad un allineamento esatto (Smith-Waterman) su una banda stretta attorno alla diagonale di maggior similarità (solitamente banda larga attorno ai 32 residui)
 su una banda stretta attorno alla diagonale di maggior similarità (solitamente banda larga attorno ai 32 residui)")
74
Sequence similarity with FASTA

75
BLAST Data un data base di sequenze, questo viene indicizzato: per ogni tripletta di residui consecutivi si memorizza in quali sequenze e in quali posizioni questa tripletta viene trovata. AAA AAC AAD ACA ...

76
BLAST Data una sequenza (Query), viene divisa in “parole” lunghe W (generalmente W = 3 per proteine) LSHLPTLPLRLDPTNMVFGHLRI LSH, SHL, HLP, LPT, PTL, TLR, …,…, Per ognuna vengono generate le parole affini secondo la BLOSUM62: parole con punteggio > T (T = ) LSH 16 ISH 14 MSH 14 VSH 13 LAH 13 LTH 13 LNH 13
 LSH 16. ISH 14. MSH 14. VSH 13. LAH 13. LTH 13. LNH 13.")
77
BLAST Per ognuna delle parole affini vengono recuperate le sequenze del data base che la contengono (secondo l’indicizzazione) La corrispondenza viene estesa (senza gap) a destra e a sinistra fino a che lo score rimane superiore a una soglia S
 a destra e a sinistra fino a che lo score rimane superiore a una soglia S.")
78
Sequence similarity with BLAST (Basic Local Alignment Search Tool)
")
79
FASTA

81
BLAST

83
Allineamento di tutte le sequenze
ATTENZIONE: Non è allineamento multiplo ottimale

84
Profilo di sequenza

85
Utilità del profilo di sequenza
Il profilo di sequenza dà una descrizione complessiva di tutte le sequenze: evidenzia le zone più conservate o le mutazioni più frequenti posizione per posizione Allineare una sequenza contro un profilo I parametri di un allineamento sono generalmente identici per tutte le posizioni. Allineare contro un profilo pesa differentemente le mutazioni nelle differenti posizioni

86
Profilo delle sequenze rintracciate
PSI-BLAST Sequenza BLAST Profilo delle sequenze rintracciate Data Base PSI-BLAST Fino a convergenza

87
The design of PSI-BLAST
PSI-BLAST takes as an input a single protein sequence and compares it to a protein database, using the gapped BLAST program The program constructs a multiple alignment, and then a profile, from any significant local alignments found. The original query sequence serves as a template for the multiple alignment and profile, whose lengths are identical to that of the query. Different numbers of sequences can be aligned in different template positions The profile is compared to the protein database, again seeking local alignments. After a few minor modifications, the BLAST algorithm can be used for this directly. PSI-BLAST estimates the statistical significance of the local alignments found. Because profile substitution scores are constructed to a fixed scale, and gap scores remain independent of position, the statistical theory and parameters for gapped BLAST alignments remain applicable to profile alignments. Finally, PSI-BLAST iterates, by returning to step (2), an arbitrary number of times or until convergence.
, an arbitrary number of times or until convergence.")
89
Motivi di sequenza Da allineamenti locali e considerazioni strutturali possono essere derivati motivi sequenziali importanti. Espressioni regolari Allineando i seguenti frammenti che coordinano un atomo di Zn C H C I C R I C C H C L C K I C C H C I C S L C D H C L C T I C C H C I D S I C Deriviamo la seguente espressione regolare [CD]-H-C-[IL]-[CD]-[RKST]-[IL]-C

90
Ricerca di motivi Possiamo cercare l’espressione regolare in diverse proteine [CD]-H-C-[IL]-[CD]-[RKST]-[IL]-C ..ALCPCHCLCRICPLIY.. ..KFRLCWCLCKICLKDF.. ..GGPLCHCICSLDASDQ.. ..FLPRCHCLCTICPIYL.. ..WERWDHCIDSICLKDE.. ..LPPICHCLCKICFGLK.. Yes No
![Ricerca di motivi Possiamo cercare l’espressione regolare in diverse proteine. [CD]-H-C-[IL]-[CD]-[RKST]-[IL]-C.](http://slideplayer.it/slide/978467/3/images/90/Ricerca+di+motivi+Possiamo+cercare+l%E2%80%99espressione+regolare+in+diverse+proteine.+%5BCD%5D-H-C-%5BIL%5D-%5BCD%5D-%5BRKST%5D-%5BIL%5D-C..jpg "..ALCPCHCLCRICPLIY.. ..KFRLCWCLCKICLKDF.. ..GGPLCHCICSLDASDQ.. ..FLPRCHCLCTICPIYL.. ..WERWDHCIDSICLKDE.. ..LPPICHCLCKICFGLK.. Yes. No.")
91
PROSITE

92
PROSITE

93
Problemi…. Espressioni regolari La risposta dello scan è sempre Sì o No ..ALCPCHCLCRICPLIY.. ..è riconosciuto come legante Zn, così come... ..WERWDHCIDSICLKDE.. …anche se l’occorrenza di due acidi aspartici come leganti non è mai osservata Allineamenti I parametri di sostituzione, di apertura di gap, ecc.. sono indipendenti dalla posizione, anche se in realtà esistono grossi vincoli all’interno di una stessa famiglia

Presentazioni simili

















>")



