Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
RICERCA DI SIMILARITA’ IN BANCHE DATI
Una sequenza “da sola” non e’ informativa, è utile poterla confontare alle sequenze note nei database perche’ possano essere formulate delle ipotesi sulla sue relazioni evolutive con sequenze simili o sulla sua funzione. Metodi di ricerca di similarità in banca dati: programmi che permettono di fare lo “screening” di una banca dati usando una sequenza “sonda”/”esca” (detta query) come input ( le sequenze nel DB sono chiamate subject) Devono essere veloci, selettivi e sensibili Si basano su metodi euristici Utilizzano allineamenti locali per confrontare le sequenze Algoritmo “Euristico” = in matematica e informatica un particolare tipo di algoritmo la cui soluzione non è la soluzione ottima per quel dato problema ma una soluzione approssimativamente molto vicina a quella ottima con tempi di calcolo ragionevoli.
 come input ( le sequenze nel DB sono chiamate subject) Devono essere veloci, selettivi e sensibili. Si basano su metodi euristici. Utilizzano allineamenti locali per confrontare. le sequenze. Algoritmo Euristico = in matematica e informatica un particolare tipo di algoritmo la cui soluzione non è la soluzione ottima per quel dato problema ma una soluzione approssimativamente molto vicina a quella ottima con tempi di calcolo ragionevoli.")
2
Se due sequenze hanno una identità > 30%:
Sono strutturalmente e di solito anche funzionalmente simili Hanno un antenato in comune Se due sequenze hanno una identità 15 < X < 30 %: Hanno un ripiegamento globale simile Siamo nella “twilight zone” Se esiste una vera relazione di solito si trovano “icebergs” (regioni di similarità locali)
")
3
Ricerche di similarità in banche dati

5
BLAST (Basic Local Alignment Search Tool)
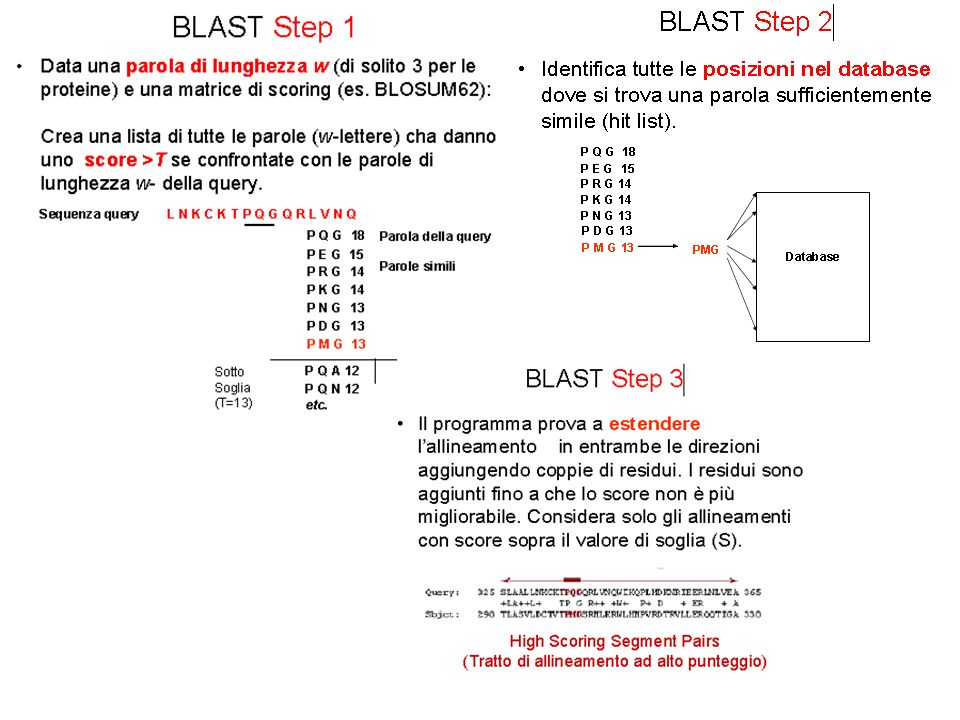
1STEP: Divisione della sequenza in parole di x caratteri (default x=3) Esempio se x=2 PAROLE POSSIBILI: AC, CD, DD, DE, EF, FG, GS, SA, AT, TR, RM, MA, AS, ST, RK 2 STEP: generazione di una lista di parole “affini” (W-mers) simili alle parole della query usando i punteggi di similarità una matrice di sostituzione selezionata dall’utente
 Esempio se x=2 PAROLE POSSIBILI: AC, CD, DD, DE, EF, FG, GS, SA, AT, TR, RM, MA, AS, ST, RK. 2 STEP: generazione di una lista di parole affini (W-mers) simili alle parole della query usando i punteggi di similarità una matrice di sostituzione selezionata dall’utente.")
6
BLAST (Basic Local Alignment Search Tool)
Sono considerati tutti i W-mers (parole affini) che superano una soglia fissata T quando allineati con parole della QUERY 3. Vengono esaminate tutte le sequenze subject, per cercare la presenza di tutti i W-mers dell’elenco. Ogni corrispondenza trovata (hit) viene considerata come parte di un allineamento più esteso. Si considera la possibilità di estendere ogni hit in ambo le direzioni. Si ottiene un segmento di allineamento locale detto HSP (high scoring segment pair). Si estendono i match cercando regioni di alta similarità fintanto che lo score è sopra un secondo valore di soglia S, fermandosi quando lo score non può più essere migliorato (lo score finale è chiamato opt)
 che superano una soglia fissata T quando allineati con parole della QUERY. 3. Vengono esaminate tutte le sequenze subject, per cercare la presenza di tutti i W-mers dell’elenco. Ogni corrispondenza trovata (hit) viene considerata come parte di un allineamento più esteso. Si considera la possibilità di estendere ogni hit in ambo le direzioni. Si ottiene un segmento di allineamento locale detto HSP (high scoring segment pair). Si estendono i match cercando regioni di alta similarità fintanto che lo score è sopra un secondo valore di soglia S, fermandosi quando lo score non può più essere migliorato (lo score finale è chiamato opt)")
8
Valutazione significatività dei match identificati
Quanto il match (query vs seq x del DB) identificato dagli allineamenti locali di BLAST è significativo? Tanto più il loro allineamento è diverso da uno generato casualmente tra sequenze di lunghezza paragonabile Sequenze che danno un allineamento casuale: –Sequenze non omologhe –Sequenze rimescolate(“shuffled”) –Sequenze generate casualmente –Sequenze a bassa complessità (con regioni ricche di amminoacidi dello stesso tipo altamente ripetuti) N.B. Blast permette di mascherare le regioni di sequenza a bassa complessitè
 identificato dagli allineamenti locali di BLAST è significativo Tanto più il loro allineamento è diverso da uno generato casualmente tra sequenze di lunghezza paragonabile. Sequenze che danno un allineamento casuale: –Sequenze non omologhe. –Sequenze rimescolate( shuffled ) –Sequenze generate casualmente. –Sequenze a bassa complessità (con regioni ricche di amminoacidi dello stesso tipo altamente ripetuti) N.B. Blast permette di mascherare le regioni di sequenza a bassa complessitè.")
9
Valutazione significatività dei match identificati
Generazione di un ampio numero di sequenze casuali con la stessa composizione della query (sequenze shuffled) Ripetizione della ricerca di similarita’su sottoinsiemi casuali delDB utilizzando come query ciascuna delle seq. casuali Calcolo dei punteggi di allineamento corrispondenti (opt) DISTRIBUZIONE DEI PUNTEGGI CASUALI Score significativi Due sequenze possono essere considerate omologhe se il punteggio per il loro allineamento ottimale (opt) cade fuori dalla distribuzione dei punteggi ottenuti per caso
 Ripetizione della ricerca di similarita’su sottoinsiemi casuali delDB utilizzando come query ciascuna delle seq. casuali. Calcolo dei punteggi di allineamento corrispondenti (opt) DISTRIBUZIONE DEI PUNTEGGI CASUALI. Score significativi. Due sequenze possono essere considerate omologhe se il punteggio per il loro allineamento ottimale (opt) cade fuori dalla distribuzione dei punteggi ottenuti per caso.")
10
E-value e Z-score Z-score esprime il numero di deviazioni standard che separano il punteggio (opt) della query della media dei punteggi casuali Z-score >= 4 Z-score= (optquery–Mcasuale) / σcasuale E-value= expectation value, numero atteso di sequenze che danno per caso il punteggio opt Indica quanto e’ probabile che si trovi il punteggio S per caso in una distribuzione di Poisson con valore medio Mcasuale NB IN BLAST il punteggio OPT puo’ essere convertito in scala logaritmica al punteggio cosidetto BIT Indicazioni: opt/bit elevati, Z-score >=4, Evalue prossimo a 0
 / σcasuale. E-value= expectation value, numero atteso di sequenze che danno per caso il punteggio opt. Indica quanto e’ probabile che si trovi il punteggio S per caso in una distribuzione di Poisson con valore medio Mcasuale. NB IN BLAST il punteggio OPT puo’ essere convertito in scala logaritmica al punteggio cosidetto BIT. Indicazioni: opt/bit elevati, Z-score >=4, Evalue prossimo a 0.")
11
E-value e Z-score Z-score= (optquery–Mcasuale) / σcasuale
Z-score esprime il numero di deviazioni standard che separano il punteggio (opt) della query della media dei punteggi casuali Z-score >= 4 Z-score= (optquery–Mcasuale) / σcasuale E-value= expectation value, numero atteso di sequenze che danno per caso il punteggio opt Indica quanto e’ probabile che si trovi il punteggio S per caso in una distribuzione di Poisson con valore medio Mcasuale NB IN BLAST il punteggio OPT puo’ essere convertito in scala logaritmica al punteggio cosidetto BIT
 della query della media dei punteggi casuali. Z-score >= 4. Z-score= (optquery–Mcasuale) / σcasuale. E-value= expectation value, numero atteso di sequenze che danno per caso il punteggio opt. Indica quanto e’ probabile che si trovi il punteggio S per caso in una distribuzione di Poisson con valore medio Mcasuale. NB IN BLAST il punteggio OPT puo’ essere convertito in scala logaritmica al punteggio cosidetto BIT.")
12
BLAST blastp cerca in database di sequenze proteiche usando come query sequenze proteiche blastn cerca in un database di sequenze nucleotidiche usando come query sequenze nucleotidiche blastx cerca in un database di sequenze proteiche partendo da una sequenza query nucleotidica che viene tradotta in tutti i frame tblastn cerca in un database di sequenze nucleotidiche partendo da una sequenza query proteica – le seq subject del database sono tradotte in sequenze proteiche in tutti i frame PSI-Blast ricerca iterativa con PSI-Blast usando ad ogni iterazione una sequenza consenso derivata dall’allineamento tra la seq query le le subject dell’interazione precedente

13
Strategie per la ricerca di similarità di proteine
Utilizzare una matrice tipo BLOSUM62 o PAM120 e la penalità per i gap di default Se non si trovano risultati significativi utilizzare BLOSUM30 o PAM250 e una penalità più bassa per i gap Prendere in considerazione i risultati con un grado di significatività tra EXP 0.05 e 10. Utilizzare PSI-BLAST per la ricerca di famiglie di proteine

14
Ricerca di pattern e motivi funzionali
Uno dei primi scopi della biologia computazionale consiste nel rispondere alla domanda: data una nuova sequenza, cosa si può dire sulla funzione (o funzioni) in essa codificata? Se la ricerca per similarità non fornisce risposte, o se occorrono ulteriori informazioni, si devono usare altri strumenti: la ricerca di pattern e motivi funzionali La pattern recognition è un’area di ricerca dell’informatica e della fisica applicata con ricadute in campi quali l’intelligenza artificiale, la linguistica computazionale, le scienze cognitive, la statistica matematica, ecc. Studia l’organizzazione e il disegno di sistemi che riconoscano motivi e regolarità nei dati a disposizione
 in essa codificata Se la ricerca per similarità non fornisce risposte, o se occorrono ulteriori informazioni, si devono usare altri strumenti: la ricerca di pattern e motivi funzionali. La pattern recognition è un’area di ricerca dell’informatica e della fisica applicata con ricadute in campi quali l’intelligenza artificiale, la linguistica computazionale, le scienze cognitive, la statistica matematica, ecc. Studia l’organizzazione e il disegno di sistemi che riconoscano motivi e regolarità nei dati a disposizione.")
15
Espressioni regolari – motivi di tipo deterministico
Un motivo (pattern) è un insieme di caratteri (nucleotidi o aminoacidi), non necessariamente contigui, associati spesso ad una precisa struttura o funzione La loro esistenza dipende dal fatto che l’evoluzione ha prodotto pochi modi per realizzare una determinata funzione I motivi possono essere codificati in: Espressioni regolari – motivi di tipo deterministico Forme più complesse (allineamenti, profili, HMM, matrici, consensus) Motivo ideale può essere sempre ed univocamente associato ad una precisa struttura o funzione Motivo reale si trova in sequenze che non presentano la funzione (falsi positivi) o è assente in sequenze funzionalmente correlate al motivo (falsi negativi) Le espressioni regolari sono formule che si possono usare per definire pattern testuali utilizzando le lettere dell’alfabeto e meta-caratteri <$+*[{( )?. ai quali è associata una determinata funzione
 è un insieme di caratteri (nucleotidi o aminoacidi), non necessariamente contigui, associati spesso ad una precisa struttura o funzione. La loro esistenza dipende dal fatto che l’evoluzione ha prodotto pochi modi per realizzare una determinata funzione. I motivi possono essere codificati in: Espressioni regolari – motivi di tipo deterministico. Forme più complesse (allineamenti, profili, HMM, matrici, consensus) Motivo ideale può essere sempre ed univocamente associato ad una precisa struttura o funzione. Motivo reale si trova in sequenze che non presentano la funzione (falsi positivi) o è assente in sequenze funzionalmente correlate al motivo (falsi negativi) Le espressioni regolari sono formule che si possono usare per definire pattern testuali utilizzando le lettere dell’alfabeto e meta-caratteri. <$+*[{( ) . ai quali è associata una determinata funzione.")
16
BANCHE DATI DI MOTIVI All’interno di un singolo motivo l’informazione può essere ridotta a una SEQUENZA CONSENSO che non deve essere necessariamente stringente: PATTERN; Se ci si riferisce a un gruppo di motivi conservati non contigui nella sequenza: FINGERPRINT oppure BLOCCHI; Se invece non si identificano regioni locali di similarità tra proteine di una stessa famiglia ma l’informazione viene dal considerare la similarità lungo l’intero allineamento si ha un PROFILO

17
MOTIVI…E MOTIVI PATTERN FINGERPRINT O BLOCCHI
PROFILO : possibilità di ricavare una sequenza consenso per tutto l’allineamento XXXXhhhhXXXbbxxaaxxNGG(X)5-8SWXX…
5-8SWXX…")
18
Ricerca di pattern e motivi funzionali in sequenze proteiche
Dallo studio di allineamenti multipli di sequenze appartenenti ad una stessa famiglia di proteine, appare evidente che alcune regioni sono più conservate regioni importanti per la funzione o la struttura Le regioni più conservate sono in genere quelle più importanti per la funzione Dalle regioni costanti e variabili di un multi-allineamento di proteine omologhe derivare un pattern che serva a distinguerle, cioè si può identificare un motivo che possa servire alla caratterizzazione funzionale delle proteine che lo contengono. Esistono diversi programmi per l’individuazione di motivi: PROSITE BLOCKS, PRINTS (fingerprint= insieme di piu’ motivi) E di domini: PFAM, SMART
 E di domini: PFAM, SMART.")
19
Le proteine ed i domini proteici che appartengono ad una particolare famiglia generalmente condividono attributi funzionali e derivano da un “antenato” comune. Dallo studio di sequenze risulta evidente che alcune regioni si conservano meglio di altre nel corso dell’evoluzione. Queste regioni in genere sono importanti per il mantenimento della struttura tridimensionale o per la funzione di una proteina. Analizzando le proprietà che vengono mantenute costanti e quelle che invece variano è possibile ottenere una “signature” per ogni famiglia proteica o dominio che consente di distinguere i suoi membri dalle altre proteine non correlate.

20
BANCHE DATI DI MOTIVI All’interno di un singolo motivo l’informazione può essere ridotta a una SEQUENZA CONSENSO che non deve essere necessariamente stringente: PATTERN PROSITE Se ci si riferisce a un gruppo di motivi conservati non contigui nella sequenza: FINGERPRINT oppure BLOCCHI BLOCKS PRINTS Se invece non si identificano regioni locali di similarità tra proteine di una stessa famiglia ma l’informazione viene dal considerare la similarità lungo l’intero allineamento si ha un PROFILO Prosite Pfam

21
MOTIVI…E MOTIVI PATTERN FINGERPRINT O BLOCCHI
PROFILO : possibilità di ricavare una sequenza consenso per tutto l’allineamento XXXXhhhhXXXbbxxaaxxNGG(X)5-8SWXX…
5-8SWXX…")
22
Database of protein families and domains
PROSITE Database of protein families and domains PROSITE è un database di famiglie proteiche (motivi associati a funzione) Consiste di siti, pattern e profili che aiutano ad identificare a quale famiglia di proteine note (se esiste) appartenga una nuova sequenza. Questo database si basa sull’osservazione che, nonostante esista un numero considerevole di proteine, la maggior parte di esse possono essere raggruppate in un numero limitato di famiglie a seconda del livello di similarità tra le sequenze. I motivi possono essere codificati in due modi: Pattern – riconducibili ad espressioni regolari Matrici (o profili) – riferiti a l’intera sequenza/domini
 Consiste di siti, pattern e profili che aiutano ad identificare a quale famiglia di proteine note (se esiste) appartenga una nuova sequenza. Questo database si basa sull’osservazione che, nonostante esista un numero considerevole di proteine, la maggior parte di esse possono essere raggruppate in un numero limitato di famiglie a seconda del livello di similarità tra le sequenze. I motivi possono essere codificati in due modi: Pattern – riconducibili ad espressioni regolari. Matrici (o profili) – riferiti a l’intera sequenza/domini.")
23
E’ un database di famiglie e domini proteici comprensiva di pattern e
motivi (signatures) che identificano e rendono riconoscibili e classificabili le proteine una signature formattata, definita anche pattern.
 che identificano e rendono riconoscibili e. classificabili le proteine. una signature formattata, definita anche pattern.")
24
PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali)
Es. phosphorylation by CK2 [ST]-x(2)-[DE]
-[DE]")
25
POTERE DIAGNOSTICO DEL PATTERN
La sensibilità di un motivo è la misura della proporzione di sequenze della famiglia selezionate dal motivo ed è data da: Sensibilità = VP/(VP + FN) se FN=0 sensibilità=1 Dove VP= veri positivi e FN=falsi negativi La selettività di un motivo misura la proporzione di sequenze della famiglia sul totale delle sequenze selezionate ed è data da: Selettività= VP/(VP + FP) se FP=0 selettività=1 Dove FP= falsi positivi
 se FN=0 sensibilità=1. Dove VP= veri positivi e FN=falsi negativi. La selettività di un motivo misura la proporzione di sequenze della famiglia sul totale delle sequenze selezionate ed è data da: Selettività= VP/(VP + FP) se FP=0 selettività=1. Dove FP= falsi positivi.")
26
Specificità = VN/(VN + FP)
La specificità misura la proporzione di sequenze che non fanno parte della famiglia, ma non vengono selezionate dal motivo ed è data da: Specificità = VN/(VN + FP) Dove VN=veri negativi e FP=falsi positivi
 Dove VN=veri negativi e FP=falsi positivi.")
27
PROSITE: pattern funzionali – pattern relativi a corte seq con alta probabilità di accadimento (modificazioni e non funzioni) – profili – indicazioni su SPECIFICITA’ e SENSIBILITA’ del pattern (POTERE DIAGNOSTICo); PRINTS: costruisce la signature di una famiglia – si autoimplementa usando SwissProt partendo da un allineamento seme – punteggi basati sulla freq di vedere un aa in una posizione BLOCKS: parte dalle famiglie di Prosite – punteggi dati con Blosum 62 – ogni blocco valutato sulla base di 2 punteggi: livello a cui 99,5% sono negativi e forza della ricerca (devono essere ben distinti e la forza maggiore del primo punteggio) Pfam: fornisce info più strutturali – entry: famiglie (in comune più domini) oppure domini – ci sono poi repeats, sequenze segnale, regioni transmembrana, regioni a bassa complessità
 Pfam: fornisce info più strutturali – entry: famiglie (in comune più domini) oppure domini – ci sono poi repeats, sequenze segnale, regioni transmembrana, regioni a bassa complessità.")
28
Pfam è una raccolta di proteine allineate e di profili generati con gli HMM (Hidden Markov Models) che descrivono quasi tutte le famiglie e i domini proteici conosciuti. Permette di ottenere informazioni strutturali e la composizione in domini di una sequenza

29
Profilo= rappresentazione numerica dell’allineamento che valuta:
Quali residui sono presenti in ogni posizione e con quale frequenza. Presenza di residui assolutamente conservati. Presenza di eventuali gap. Dagli allineamenti multipli si possono costruire delle position-specific scoring tables (PSST), che riflettono la probabilità di trovare un certo aminoacido in ogni posizione.
, che riflettono la probabilità di trovare un certo aminoacido in ogni posizione.")
30
METODO DEI PROFILI ALLINEAMENTO MULTIPLO di seq OMOLOGHE: vedo come seq libera di variare nel corso dell’evoluzione; Metodo dei profili permette analisi della VARIABILITA’ dei residui in una certa posizione per una certa famiglia Costruzione di MATRICI POSIZIONE-DIPENDENTI Un profilo è una matrice nX20 dove n è la posizione nell’allin.multiplo

31
I programmi che partendo da un allineamento multiplo generano i profili riportano la sequenza consenso derivante dagli allineamenti - la sequenza consenso mi da’ informazioni sui residui piu’ frequenti in quelle posizioni dell’allineamento

32
Il profilo può essere utilizzato per una ricerca in banca dati mediante la variante di Blast PSI-BLAST. Il programma effettua, data una sequenza query, una serie di iterazioni in cui ogni volta l’utente sceglie un certo numero di sequenze individuate e su queste viene ricostruito il profilo. Ad ogni iterazione successiva verranno individuate nuove sequenze, in modo più o meno accurato a seconda delle scelte fatte in precedenza.

Presentazioni simili



 e nel verificare se con i dati a disposizione è possibile rifiutarla o no.>")







>")








