Scaricare la presentazione
1
IPOSOMIA G. Chiumello Centro di Endocrinologia dell’ Infanzia e dell’ Adolescenza, Università Vita-Salute San Raffaele, Milano

2
DEFINIZIONE DI BASSA STATURA
statura < al 3° centile per le tabelle di Tanner statura < alle - 2 DS per le tabelle di Tanner statura < alle - 2 DS rispetto al target genetico velocità di crescita inferiore al 25° centile o a –1DS (‘scarso accrescimento’)
")
3
CLASSIFICAZIONE Bassa statura familiare VARIANTI NORMALI
Ritardo costituzionale di crescita e pubertà Osteocondrodisplasie DISARMONICHE Rachitismi VARIANTI PATOLOGICHE Cromosomopatie Sd dismorfiche IUGR PRENATALI Iponutrizione Malassorbimento Mal. Metaboliche Mal. Cardiache Mal. Polmonari Mal. Renali Epatopatie Emopatie Mal. Infiammatorie croniche Endocrinopatie ARMONICHE POSTNATALI

4
APPROCCIO DIAGNOSTICO
Note anamnestiche gravidanza e periodo perinatale dati auxologici neonatali (SGA) familiarità per ipostaturalismo/ritardo di sviluppo puberale segni/sintomi associati malattie croniche o pregresse assunzione di farmaci (steroidi) alimentazione, condizione socioeconomica problemi comportamentali
 familiarità per ipostaturalismo/ritardo di. sviluppo puberale. segni/sintomi associati. malattie croniche o pregresse. assunzione di farmaci (steroidi) alimentazione, condizione socioeconomica. problemi comportamentali.")
5
DATI AUXOLOGICI percentile di crescita (< al 3° centile o alle - 2 DS per le tabelle di riferimento) potenziale genetico (statura <- 2 DS rispetto al target parentale) velocità di crescita (< 25° centile o –1DS) maturazione scheletrica curva ponderale
 velocità di crescita (< 25° centile o –1DS) maturazione scheletrica. curva ponderale.")
6
esame obiettivo completo
valutazione sindromologica (se opportuno) ematochimici generali indici nutrizionali, EMA, AbTG funzionalità tiroidea cariotipo (femmine) ulteriori indagini endocrinologiche
 ematochimici generali. indici nutrizionali, EMA, AbTG. funzionalità tiroidea. cariotipo (femmine) ulteriori indagini endocrinologiche.")
7
BASSA STATURA FAMILIARE
VARIANTI “NORMALI” BASSA STATURA FAMILIARE RITARDO COSTITUZIONALE DI CRESCITA E PUBERTA’

8
BASSA STATURA FAMILIARE
Non è una autentica patologia; i bambini che hanno un potenziale di crescita limitato da fattori ereditari presentano: deficit staturale isolato normale sviluppo puberale familiarità per bassa statura (almeno un genitore) scarto staturale correlabile con il target genetico curva di crescita inferiore ma parallela al 3° centile maturazione ossea compatibile con l’età cronologica/poco ritardata statura adulta adeguata al target genetico
 scarto staturale correlabile con il target genetico. curva di crescita inferiore ma parallela al 3° centile. maturazione ossea compatibile con l’età cronologica/poco ritardata. statura adulta adeguata al target genetico.")
9
TH . Bassa statura familiare

10
RITARDO COSTITUZIONALE DI CRESCITA E PUBERTA’
bassa statura pubertà ritardata familiarità per ritardo puberale velocità di crescita relativamente normale durante l’infanzia, picco di crescita puberale tardivo età ossea ritardata e simile all’età staturale statura finale normale nel 50% dei casi statura adulta adeguata al target genetico nel 50%

11
TH . RCCP

12
OSTEOCONDRODISPLASIE
BASSA STATURA DISARMONICA RACHITISMI

13
BASSA STATURA ARMONICA
da cause prenatali - Cromosomopatie: sd di Turner sd di Noonan sd di Down sd di Prader-Willi - Sd dismorfiche sd fetoalcolica sd di Silver-Russel - SGA

14
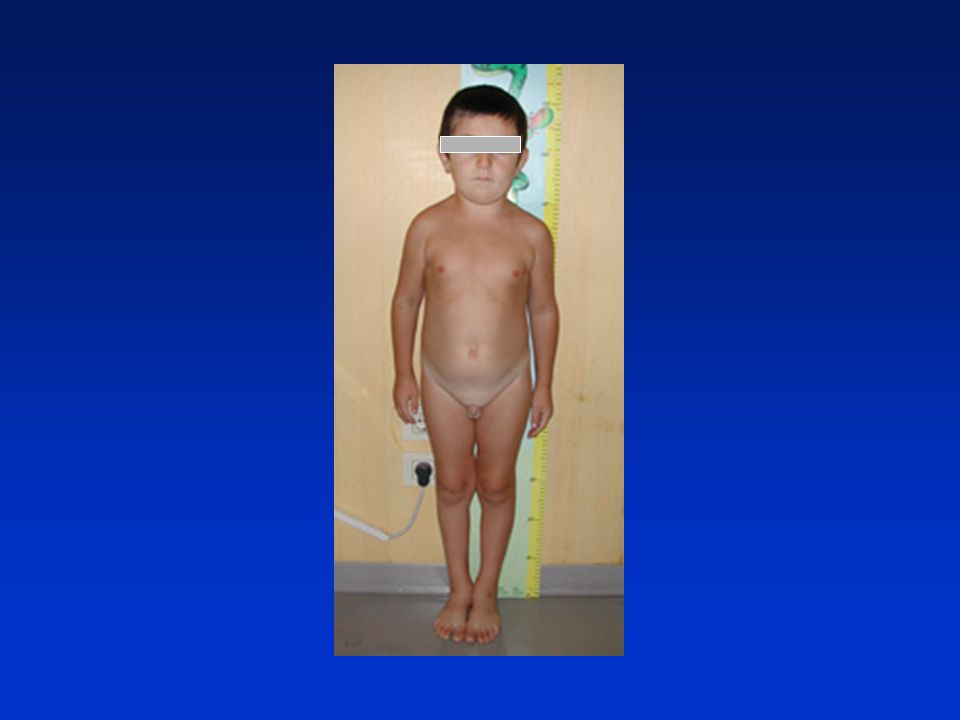
M.R. Eta’ 15 anni giunge per iposomia ed amenorrea primaria
Anamnesi: familiarità per ritardo puberale ESAME OBIETTIVO altezza 135cm, M2P2A2 ESAMI ORMONALI LH FSH 17b-estradiolo ECOGRAFIA PELVICA ipoplasia ovarica ECOGRAFIA RENALE rene a ferro di cavallo CARIOGRAMMA: XO

16
S. di TURNER Incidenza: 1:2.500 neonati di sesso femminile
Cariogramma: 45X, mosaici (46XX/45X) Fenotipo: iposomia pterigium colli impianto basso dei capelli micrognazia/ palato ogivale epicanto/ptosi palpebrale gomito valgo torace a corazza con capezzoli distanti Disgenesia gonadica: ridotta o assente produzione di E2
 Fenotipo: iposomia. pterigium colli. impianto basso dei capelli. micrognazia/ palato ogivale. epicanto/ptosi palpebrale. gomito valgo. torace a corazza con capezzoli distanti. Disgenesia gonadica: ridotta o assente produzione di E2.")
17
Turner

18
Anomalie cardiache: coartazione dell’aorta
insufficienza mitralica Anomalie renali: rene a ferro di cavallo ptosi renale malrotazione Anomalie scheletriche: brevità IV osso metacarpale disgenesia epifisaria gomito e ginocchio precoce osteoporosi Ritardo mentale: variabile

19
TERAPIA GH dose 0.05-0.06mg/kg/die guadagno staturale medio +1.5SDS
correlazione positiva con statura all’inizio del trattamento, durata della terapia, target staturale correlazione inversa con età di inizio della terapia Terapia estroprogestinica sostitutiva

20
AD INIZIO INTRAUTERINO
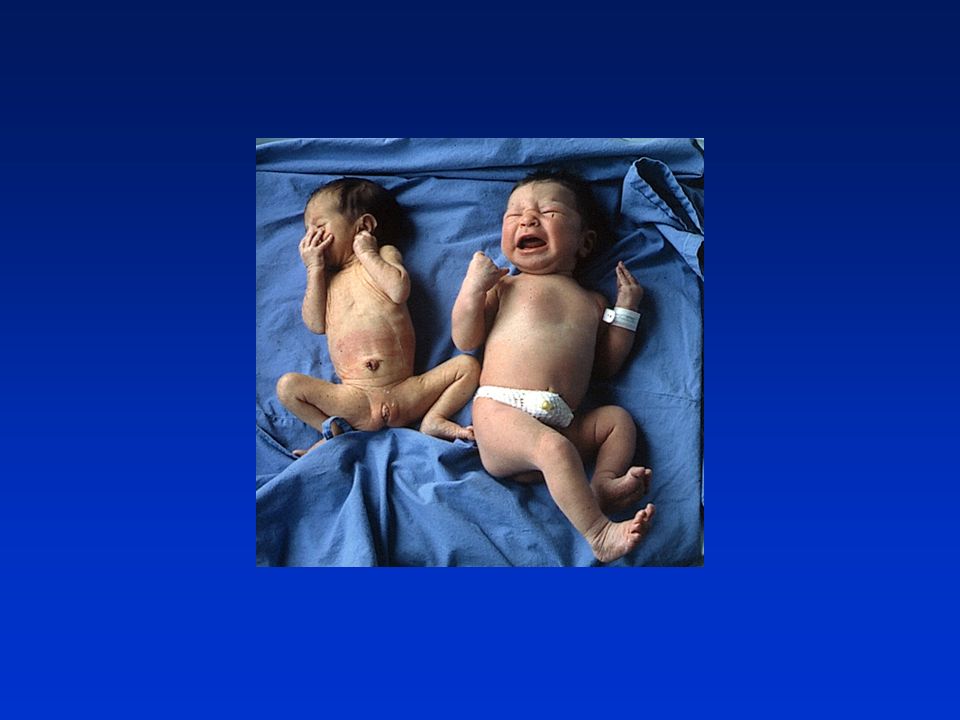
RITARDO DI CRESCITA AD INIZIO INTRAUTERINO Neonato piccolo per età gestazionale: con peso e/o lunghezza inferiore al 10°ple in relazione alla sua età gestazionale. Non tiene conto dei fattori eziopatogenetici responsabili della ridotta crescita. SGA IUGR Rallentamento di crescita intrauterino, indipendentemente dal percentile assoluto occupato alla nascita. Indica un minor potenziale di crescita fetale conseguente a processi patologici di origine materna, placentare o fetale.

21
SIMMETRICO: riduzione proporzionale di lunghezza, peso e circonferenza cranica;
se la noxa patogena si verifica nelle prime 16 settimane di vita SGA ASIMMETRICO: prevalente deficit ponderale; se la noxa patogena si verifica nel terzo trimestre

23
il 90% recupera altezza e peso entro i 2-4 anni di età
il 10% raggiunge una statura finale non soddisfacente crescono meglio i bambini con prevaente deficit ponderale alla nascita raro il riscontro di deficit di GH; patogenesi complessa

24
possibilità di trattamento con ormone della crescita nel nato SGA
IL PASSATO: possibilità terapeutica se deficit di GH IL FUTURO: possibile trattare sulla base di criteri auxologici

25
Necessarie dosi più elevate di GH rispetto al bambino con deficit di GH e peso adeguato
Aumento della velocità di crescita Miglioramento della statura adulta (anche se pochi gli studi già conclusi) Aumento della massa muscolare (più evidente nei primi 2 anni di terapia)
 Aumento della massa muscolare (più evidente nei primi 2 anni di terapia)")
26
iposviluppo fetale, TC alla 37° s.g.
D.S. iposviluppo fetale, TC alla 37° s.g. alla nascita: peso gr 1600 , lungh. 39cm, CC 34cm note dismorfiche: micrognazia, macrocefalia relativa, lieve asimmetria volto ed arti inferiori spiccata inappetenza nella prima infanzia diagnosi di s.Silver Russell

28
TH . GH

29
BASSE STATURE SECONDARIE malattie endocrine
deficit isolato di GH panipopituitarismo ipotiroidismo (tiroidite) rachitismo ipofosforemico pseudoipoparatiroidismo eccesso di glucocorticoidi sindromi da insensibilità al GH deficit primari di IGF-I
 rachitismo ipofosforemico. pseudoipoparatiroidismo. eccesso di glucocorticoidi. sindromi da insensibilità al GH. deficit primari di IGF-I.")
30
deficit di GH: epidemiologia
prevalenza 1: 6000 – 1: bambini in età scolare 1:120 bambini con bassa statura rapporto maschi/femmine è di 1: 2 spesso associati altri deficit ipofisari (70%)
")
31
deficit di GH: eziologia
idiopatico disembriogenesi ipotalamo-ipofisaria displasia setto-ottica forme con difetto genico tumori ipotalamo-ipofisari istiocitosi radiazioni traumi, emorragie, infezioni, asfissia sindromi da insensibilità al GH forme primarie forme secondarie 25%

32
TH . GH

33
peduncolo ADENOIPOFISI NEUROIPOFISI normale

34
assottigliamento peduncolo
Peduncolo assottigliato ADENOIPOFISI NEUROIPOFISI assottigliamento peduncolo

35
PSIS

36
craniofaringioma

37
deficit di GH: forme di origine genetica
delezione/mutazione del gene del GH con deficit quantitativo di GH con GH inattivo mutazione del recettore per il GHRH (raro) deficit ipofisari multipli (geni HESX1, PROP1, POU1F1) mutazione del del recettore per GH (s. Laron) difetti primari della sintesi di IGF-I difetti del recettore per IGF-I
 deficit ipofisari multipli (geni HESX1, PROP1, POU1F1) mutazione del del recettore per GH (s. Laron) difetti primari della sintesi di IGF-I. difetti del recettore per IGF-I.")
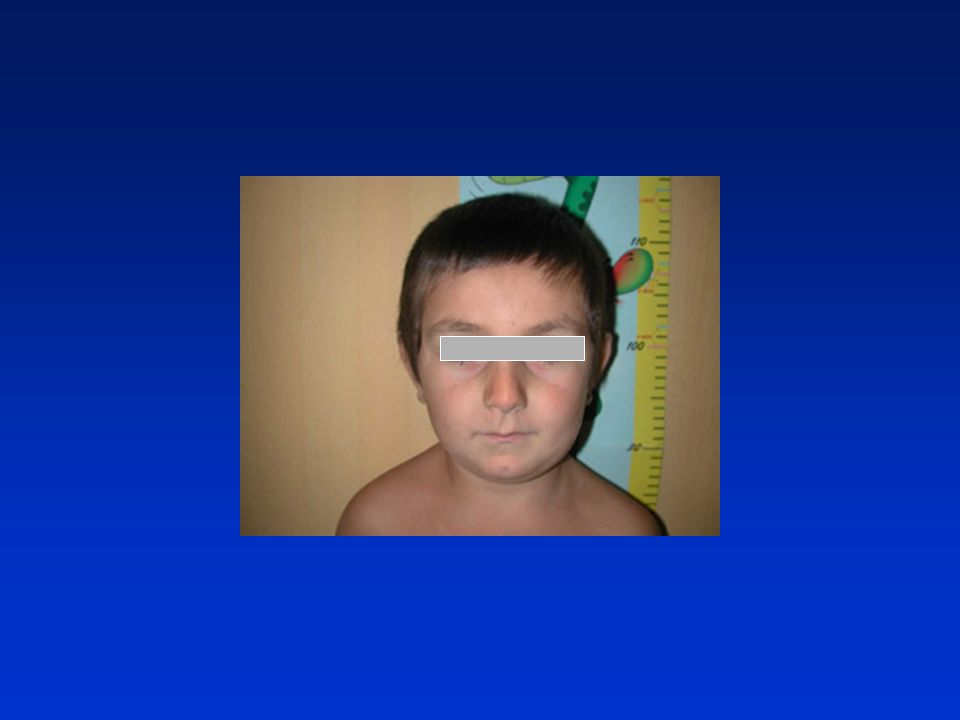
38
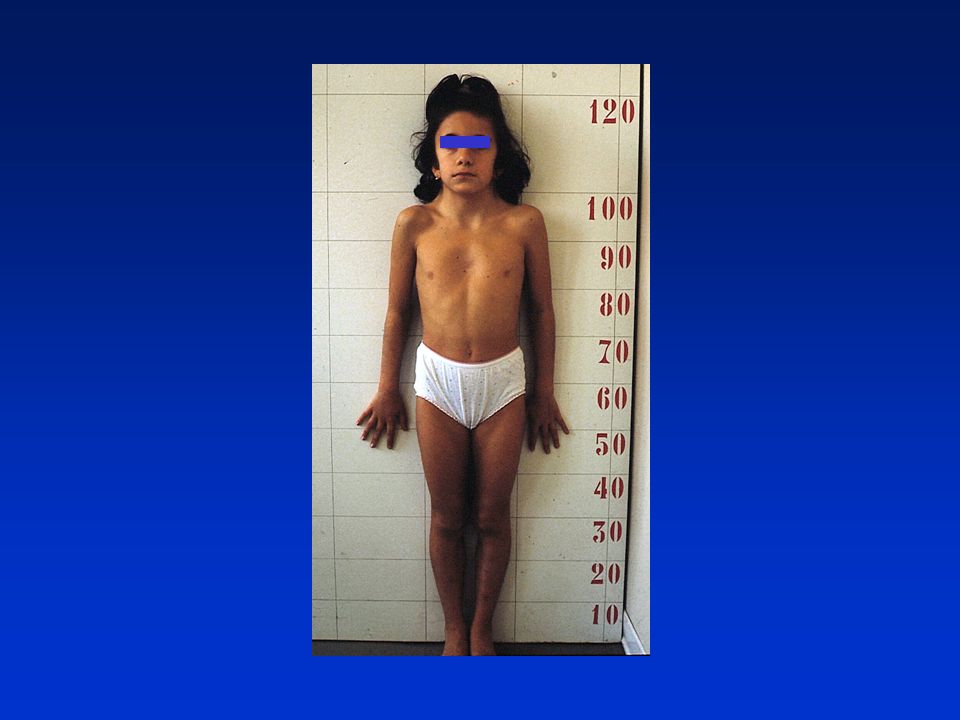
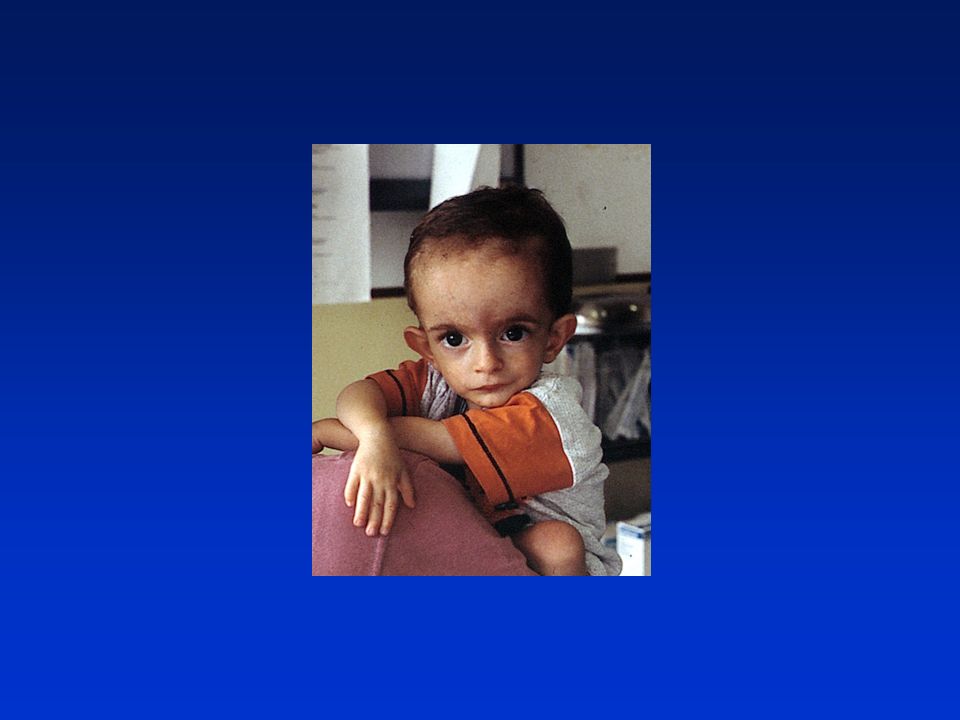
deficit di GH: aspetti clinici
bassa statura armonica aspetto infantile, voce acuta fronte ampia e bombata (facies da bambola), capelli fini naso a sella e piccolo ipoplasia modesta dello splancnocranio ritardata eruzione dentaria modesto eccesso di adipe prevalente al tronco ipoglicemia neonatale, ittero protratto, micropene sviluppo neuromotorio e intelligenza normali. età ossea molto ritardata, velocità di crescita ridotta
, capelli fini. naso a sella e piccolo. ipoplasia modesta dello splancnocranio. ritardata eruzione dentaria. modesto eccesso di adipe prevalente al tronco. ipoglicemia neonatale, ittero protratto, micropene. sviluppo neuromotorio e intelligenza normali. età ossea molto ritardata, velocità di crescita ridotta.")
41
deficit di GH: diagnosi
dati auxologici (deficit staturale, vel. di crescita) età ossea secrezione di GH ad almeno 2 test da stimolo IGF1 RM della regione ipotalamo-ipofisaria Genetica (casi selezionati)
 età ossea. secrezione di GH ad almeno 2 test da stimolo. IGF1. RM della regione ipotalamo-ipofisaria. Genetica (casi selezionati)")
42
deficit di GH: terapia ormone della crescita (dal 1985 biosintetico)
iniezioni serali sottocutanee dose standard: mg/kg/die recupero staturale ottimo nei deficit di GH classici non rilevanti effetti collaterali non confermata insorgenza di tumori/leucemie nei trattati












 in un individuo adulto in grado di procreare.>")








