Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Malattie metaboliche innate

2
Cosa sono le malattie metaboliche
Errori del metabolismo Frequenza of 1/200 o meno Detection: screening o sintomi Genetica: recessive, X-linked, mitochondriali, dominanti Possono essere trattabili o no

3
Periodo dell’insorgenza iniziale
Neonatale (<28 giorni di età) Infanzia (1 mese a 1 anno) fanciullezza Adolescenza Adulta
 Infanzia (1 mese a 1 anno) fanciullezza. Adolescenza. Adulta.")
4
Presentazione clinica
Alterazioni Neurologiche , convulsioni, coma Compromissione di diversi organi Acidosi Iperammonemia SIDS/ALTE apparent Life-Threatening Event (ALTE) and Sudden Infant Death Syndrome (SIDS): Mancato sviluppo Malformazioni Disfunzioni renali o epatiche
 and Sudden Infant Death Syndrome (SIDS): Mancato sviluppo. Malformazioni. Disfunzioni renali o epatiche.")
5
Organomegalia Anomalie ematologiche Coinvolgimento degli arti Regressione o ritardo nello sviluppo Aggressione Sintomi psichiatrici Asintomatica

6
Pensare a una malattia metabolica se :
Insorgenza: Ore a Giorni Con: Scarsa alimentazione o vomito Ipotonia / Ipertonia Letargia -- Stupore-- Coma convulsioni Apnea, Tachipnea o Iperpnea Odore inusuale (Urina, respiro, pelle, Cerume)
")
7
Approccio al neanato malato
considerare sempre la possibilità di una malattia metabolica Test iniziali: gas nel sangue, Anioni, Glucosio, Chetoni, Ammonio, Lattato Interrompere la dieta proteica Prendere il massimo delle calorie dal glucosio Test secondari : Amino Acidi nel plasma, Lattato/piruvato, Carnitina, Acidi organici nelle urine, Acido orotico

8
Test di laboratiro di base: glucosio, elettroliti
Diagnosi critica: coma,convulsioni, stress respiratorio, collasso cardiovascolare Test di laboratiro di base: glucosio, elettroliti Gas nel sangue arteioso, ammonia, urine ipoglicemia Acidosi metabolica Maggiori Anomalie Nessuna anomalia iperammonemia Chetosi

9
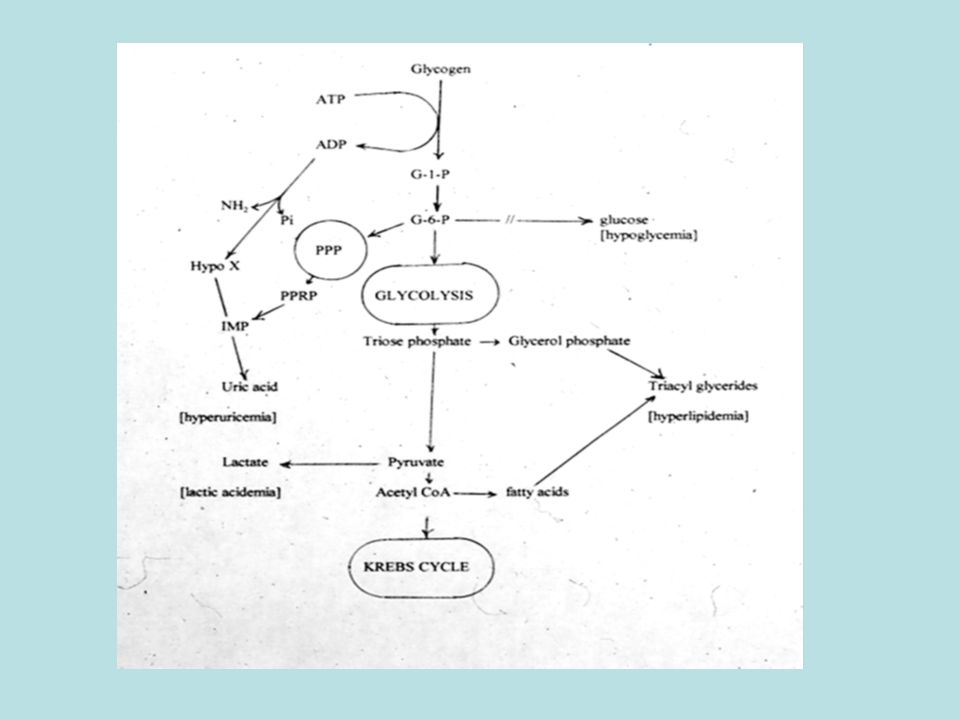
Principali anomalie Ipoglicemia Iperammonemia Ossidazione acidi grassi
(ciclo carnitina) Disordini metab. zuccheri Ipoglicemia Campioni critici sangue: insulina, GH, cortisolo 3OHB/AcAc, FFA, lattato Urina: acidi organici, Substanze riducenti Iperammonemia Difetti ciclo urea (ciclo carnitina) Campioni critici sangue: amino acidi Urina: acido orotico
 Disordini metab. zuccheri. Ipoglicemia. Campioni critici. sangue: insulina, GH, cortisolo. 3OHB/AcAc, FFA, lattato. Urina: acidi organici, Substanze riducenti. Iperammonemia. Difetti ciclo urea. (ciclo carnitina) Campioni critici. sangue: amino acidi. Urina: acido orotico.")
10
Principali anomalie Acidosi metabolica Aciduria organica
Acidosi lattica Sangue: amino acidi, lattato Urine: Acidi organici CSF (fluido cerebrospinale): lattato Campioni critici **Chetosi (DNPH) Urine a sciroppo d’acero Sangue: amino acidi Urina: Acidi organici Campioni critici Dinitrofenilidrazina (DNPH): test per la presenza di chetoacidi
: lattato. Campioni critici. **Chetosi (DNPH) Urine a sciroppo d’acero. Sangue: amino acidi. Urina: Acidi organici. Campioni critici. Dinitrofenilidrazina (DNPH): test per la presenza di chetoacidi.")
11
Principali anomalie Nessuna anomalia Organica aciduria,
Iperglicemia non chetotica, Sulfito ossidasi, Malattie da neurotrasmettitori Sangue: amino acidi, lattato Urine : acidi organici CSF: lattato, neurotrasmettitori Campioni critici

12
NKH :iperglicemia non chetotica GSD: glicogenosi
FAO: ossidazione acidi grassi MSUD: urine a scirpoo d’acero Modified from J-M Saudubray, MMBID 2001

13
CHETOSI chetosi - Acidosi metabolica + acidosi Ipoglicemia da digiuno
Glucosio normale Ipoglicemia da digiuno Chetoacidosi (vedi acidosi) Chetosi intermittente permanente +epatomegalia Iperlatticidemia Post-prandiale -epatomegalia SCAD SCHAD Dieta ricca di MCT Difetti chetolisi Gliocgenosi III,VI,IX Def. Gliocogeno sintetasi Ipoglicemia chetotica Difetti chetolisi SCHAD, MCAD Deficit glicogenosintetasi
 Chetosi. intermittente. permanente. +epatomegalia. Iperlatticidemia. Post-prandiale. -epatomegalia. SCAD. SCHAD. Dieta ricca di MCT. Difetti chetolisi. Gliocgenosi III,VI,IX. Def. Gliocogeno sintetasi. Ipoglicemia chetotica. Difetti chetolisi. SCHAD, MCAD. Deficit glicogenosintetasi.")
14
ACIDOSI GSD 1 glicogenosi tipo I PDH piruvico deidrogenasi
PC piruvico carbossilasi KGD chetoglutarato deidrogenasi MCD carbossilasi multipla TCA ciclo ac. Tricarbossilici RTA acidosi tubulare renale ACIDOSI Acidosi metabolica +chetosi -chetosi iperglicemia Alta NH3 Bassa/ norm NH3 normoglicemia ipoglicemia Lattato alto normale Diabete Difetti chetolisi Gluconeogenesi Cat. Resp. Gluocosio basso Difetto PDH (epatomegalia) Gluocosio normale Difetti: PC KGDH Difetti cat. Resp. Aciduria Organia (PA,MMA,IVA) MSUD Defetti Chetolisi Aciduria organica Deficit SCAD RTA tipo I e II MMA PA IVA Difetti Metab FA G-6-P FBP
 Gluocosio. normale. Difetti: PC. KGDH. Difetti. cat. Resp. Aciduria. Organia. (PA,MMA,IVA) MSUD. Defetti Chetolisi. Aciduria organica. Deficit SCAD. RTA. tipo I e II. MMA. PA. IVA. Difetti. Metab FA. G-6-P. FBP.")
15
ACIDEMIA LATTICA 10< >30

16
IPERAMMONEMIA citrullina

17
Malattie metaboliche specifiche
Difetti del metabolismo degli zuccheri Difetti del metabolismo degli amino acidi Difetti del metabolismo degli acidi grassi Difetti del metabolismo dei nucletidi Difetti del metabolismo energetico …………………………..

18
DIFETTI DEL METABOLISMO DEI CARBOIDRATI

19
GLICOGENOSI: difetti del metabolismo del glicogeno

20
piruvato Alanina Iperalaninemia GLICOGENOSI TIPO I:
ACETIL-CoA piruvato Glucosio-6-P GLICOGENO ACIDI GRASSI GLUCOSIO ACIDO LATTICO C. kREBS GLICOGENOSI TIPO I: DEFICIENZA DI GLUCOSIO-6-FOSFATO FOSFATASI IPOGLICEMIA IPERLATTICIDEMIA IPERLIPIDEMIA IPERURICEMIA Alanina Iperalaninemia

22
deficit dell’enzima deramificante
Glicogenosi tipo III: deficit dell’enzima deramificante GLICOGENO GLUCOSIO-6-FOSFATO Acido piruvico Acetil-CoA GLUCOSIO Acido lattico 2 3 ACIDI GRASSI CORPI CHETONICI C. kREBS 2. FOSFORILASI E FOSFORILASI CHINASI B 3. ENZIMA DERAMIFICANTE GLUCONEOGENESI CHETO- GENESI

23
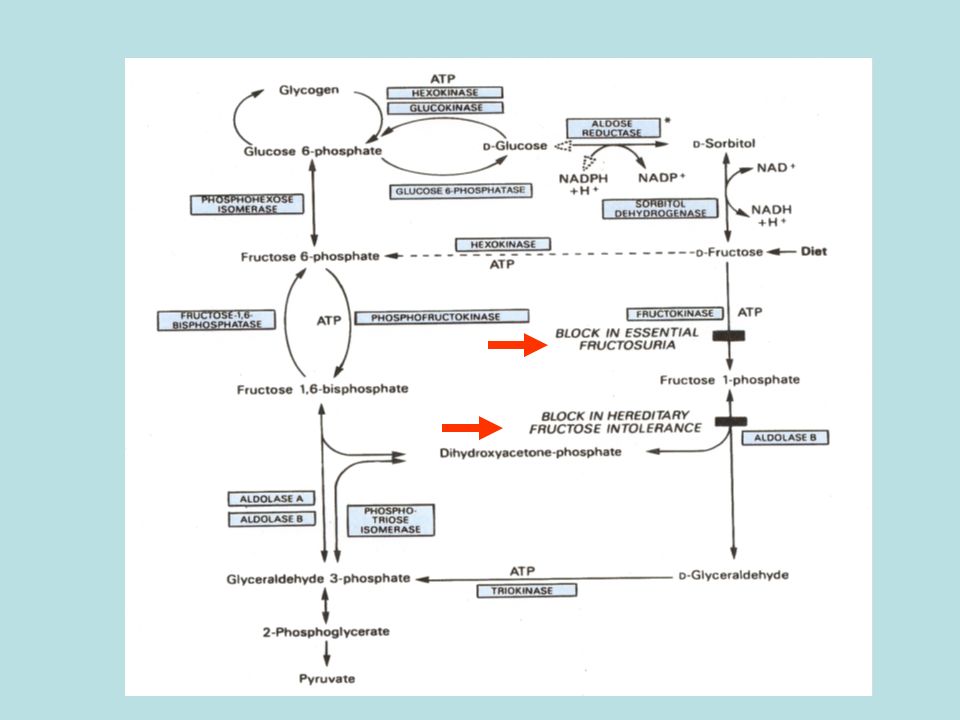
Difetti del metabolismo del fruttosio
Fruttosuria essenziale Intolleranza ereditaria al fruttosio Deficit della fruttosio 1,6-difosfatasi

25
Intolleranza ereditaria al fruttosio: deficit dell’alodolasi B
GLICOGENO G-1-P G-6-P F-6-P F-1,6-dP GAH-3-P 1,3-dPGA GAH F-1-P fruttosio sorbitolo DHA-P ATP ADP fruttochinasi Aldolasi B Blocco della glicogenolisi epatica Blocco della gluconeogenesi (Molto attiva) piruvato AMP IMP AMP deaminasi Pi GTP Acido urico GTP e Pi vengono sequestrati; Diminuisce l’inibizione su AMP deaminasi; Aumenta demolizione di IMP e Acido urico
 piruvato. AMP. IMP. AMP deaminasi. Pi. GTP. Acido urico. GTP e Pi vengono sequestrati; Diminuisce l’inibizione su. AMP deaminasi; Aumenta demolizione. di IMP e Acido urico.")
26
Difetti del metabolimso del galattosio: defcit della galattochinasi
deficit della galattosio-1-fosfato-uridiltrasferasi galattitolo Aldoso reduttasi

27
Difetti del metabolismo energetico
Difetti del metabolismo del piruvato e ciclo di Krebs Difetti dell’ossidazione degli acidi grassi Difetti della catena respiratoria

28
Difetti ossidazione acidi grassi 1 2 3 4 Acetil-CoA Corpi chetonici
CACT CPT I carnitina acilcarnitina CoASH Acil-CoA Membrana mit. Int. Membrane mit. esterna ß-osidazione CPT II Acil-CoA DH 3-OH acil-CoA DH Trasferimento di elettroni 1 2 3 4 Corpi chetonici Acetil-CoA

29
Pazienti con il deficit della traslocasi
ipoglicemia ipochetosi -iperammonemia -aciduria dicarbossilica diminuzione della carnitina libera aumento della carnitina esterificata debolezza muscolare episodi di coma indotti da digiuno aritmia cardiaca (aumento delle acilcarnitine) epatomegalia diminuzione dell’attività del trasportatore della carnitina diminuzione dell’ossidazione degli acidi grassi attività normale della CPT I e della CPT II attività normale degli enzimi di b-ossidazione
 epatomegalia. diminuzione dell’attività del trasportatore della carnitina. diminuzione dell’ossidazione degli acidi grassi. attività normale della CPT I e della CPT II. attività normale degli enzimi di b-ossidazione.")
30
Ipochetosi: scarsa prduzione di corpi chetonici
Aciduria dicarbossilica: ossidazione di alcuni acidi grassi Iperammonemia: la diminuzione di acetil CoA porta alla diminuzione di acetil-glutammato, attivatore allosterico della carbamilfosfato sintetasi Ipoglicemia: rapida diminuzione del glicogeno mancata attivazione della gluconeogenesi Il deficit della CPT II e in parte della CPT I provoca gli stessi problemi del deficit della traslocasi della carnitina

31
Diefetti del metabolimo degli amino acidi
Iperfenilalaninemie e fenilchetonuria (PKU) Tirosinemie alcaptonuria
 Tirosinemie. alcaptonuria.")
32
Metabolismo della fenilalanina e tirosina
Fenilalanina Fenilpyruvico ac. Fenilketonuria (Fenilalanina idrossilasi) Tirosina Dopa Dopamina (Tirosinasi idrossilasi) Tirosinemia II (Tirosina aminotransferasi) Paraidrossi Feenilpiruvico ac. (Paraidrossi fenilpyruvico ossidasi) Omogentisico ac. Alkaptonuria (Omogentisico acid ossidasi) Maleilacetoacetico ac Fumarilacetoacetico ac Tirosinemia I (Fumarilacetoacetato idrossilasi) Fumarico ac. + Acetoacetico ac. albinismo
 Tirosina Dopa Dopamina. (Tirosinasi idrossilasi) Tirosinemia II (Tirosina aminotransferasi) Paraidrossi Feenilpiruvico ac. (Paraidrossi fenilpyruvico ossidasi) Omogentisico ac. Alkaptonuria (Omogentisico acid ossidasi) Maleilacetoacetico ac. Fumarilacetoacetico ac. Tirosinemia I (Fumarilacetoacetato idrossilasi) Fumarico ac. + Acetoacetico ac. albinismo.")
35
Aciduria organica degli amino acidi a catena ramificata
(malattie delle urione a sciroppo d’acero) Blocco delle deidrogenasi Aciduria isovalerica (IVA) Aciduria propionica (PA) Aciduria metilmalonica (MMA)
 Blocco delle deidrogenasi. Aciduria isovalerica (IVA) Aciduria propionica (PA) Aciduria metilmalonica (MMA)")
36
Difetti del ciclo dell’urea

37
Iperammonemia Intervallo normale Neonati 64 - 107 mmol/L
Adulti mmol/L Presentazione neonatale Letargia, Anoressia, Vomito, Apnea, convulsioni, Coma, spesso fatale Infantile --ritardo sviluppo,convulsioni, Atassia, Epatomegalia, Atrofia cerebrale Stress/Febbre/

38
N Defcit conc.aa. Ac orotico
Deficienza della carbamilfosfato sintetasi glutamina N alanina citrullina arginina Deficienza della ornitina transcarbamilasi glutamina Deficienza della argininsuccinico sintetasi citrullina (citrullinemia) arginina Deficinenza della argininsuccinico liasi citrullina (aciduria argininsuccinica) acido arginsuccinico Deficienza della arginasi arginina (iperargininemia) Deficienza dellaN-acetilglutammato glutamina Sintetatsi Alanina N
 arginina. Deficinenza della argininsuccinico liasi citrullina. (aciduria argininsuccinica) acido arginsuccinico. Deficienza della arginasi arginina. (iperargininemia) Deficienza dellaN-acetilglutammato glutamina. Sintetatsi Alanina. N.")
39
Riutilizzazione e catabolismo delle basi puriniche e pirimidiniche
blue-catabolismo rosso-vie di recupero

40
Disordini del metabolismo delle Purine
Sindrome di Lesch-Nyhan Deficiencineza della Ipoxantina-Guanina Fosforibosiltransferasi (HGPRT) Accumulo di acido urico Sintomi SNC: movementi incontrollati, spasticità, self-mutilazione Deficienza dell’Adenosina Deaminasi (ADA) Perdita della funzione delle cellule B e T; grave compromissione del sistema immunitario
 Accumulo di acido urico. Sintomi SNC: movementi incontrollati, spasticità, self-mutilazione. Deficienza dell’Adenosina Deaminasi (ADA) Perdita della funzione delle cellule B e T; grave compromissione del sistema immunitario.")
41
Disordini del metabolismo delle Purine
Lesch-Nyhan Syndrome: Severe HGPRT deficiency In addition to symptoms of gout, patients display severe behavioral disorders, learning disorder, aggressiveness and hostility, including self-directed. Patients must be restrained to prevent self-mutilation. Reason for the behavioral disorder is unknown. X-linked trait (HGPRT gene is on X chromosome). Severe combined immune deficiency (SCID): lack of adenosine deaminase (ADA). Lack of ADA causes accumulation of deoxyadenosine. Immune cells, which have potent salvage pathways, accumulate dATP, which blocks production of other dNTPs by its action on ribonucleotide reductase. Immune cells can’t replicate their DNA, and thus can’t mount an immune response.
. Severe combined immune deficiency (SCID): lack of adenosine deaminase (ADA). Lack of ADA causes accumulation of deoxyadenosine. Immune cells, which have potent salvage pathways, accumulate dATP, which blocks production of other dNTPs by its action on ribonucleotide reductase. Immune cells can’t replicate their DNA, and thus can’t mount an immune response.")
42
ADA (adenosine deaminase)
Catalizza la deaminazione di adenosina e desossiadenosina in inosina e desossiinosina Mutazioni nel gene dell’ADA causano immunodeficienza (SCID, Severe Combined Immunodeficiency): virtualmente assente attivita’ di linfociti B e T Cure possibili: trapianto di midollo, somministrazione di PEG-ADA bovina
: virtualmente assente attivita’ di linfociti B e T. Cure possibili: trapianto di midollo, somministrazione di PEG-ADA bovina.")
43
Trial clinico per deficit ADA

44
Trial clinico per deficit ADA
L’espressione di ADA persiste a 12 anni dalla prima infusione di cellule modificate in una delle 2 pazienti trattate: 20% dei linfociti esprimono ancora ADA La 2a paziente ha invece sviluppato anticorpi contro componenti del sistema di infezione (FCS) e ha perso i linfociti trasdotti e l’espressione di ADA
 e ha perso i linfociti trasdotti e l’espressione di ADA.")
45
Restraints used to prevent self-mutilation in Lesch-Nyhan
patients.

46
HGPRT Deficiency

47
Mitochondrial Disorders

Presentazioni simili

 DIAGNOSI GENETICA PREIMPIANTO ( DGP )>")


















