Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
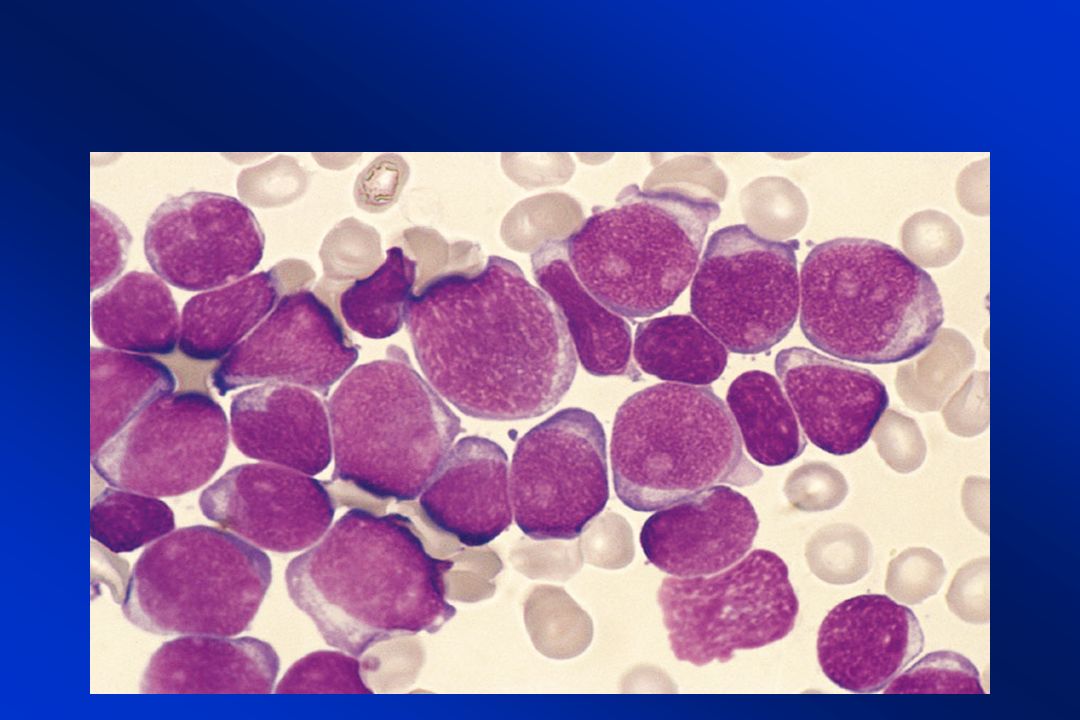
Case Report A 26-year-old woman was referred for progressive fatigue, dyspnea, blurred vision, and near- syncope. At presentation, she had marked pallor, pyrexia, diffuse nontender lymphadenopathy, and both preretinal and retinal hemorrhages. The initial laboratory examination revealed a leukocyte count of 249,000/µL, a hemoglobin concentration of 2.9 mg/dL (normal range, 11.5 to 16.0), and a platelet count of 48,000/µL. Mauro MJ. N Engl J Med 2003;349:767
, and a platelet count of 48,000/µL. Mauro MJ. N Engl J Med 2003;349:767.")
2
Mauro MJ. N Engl J Med 2003;349:767

4
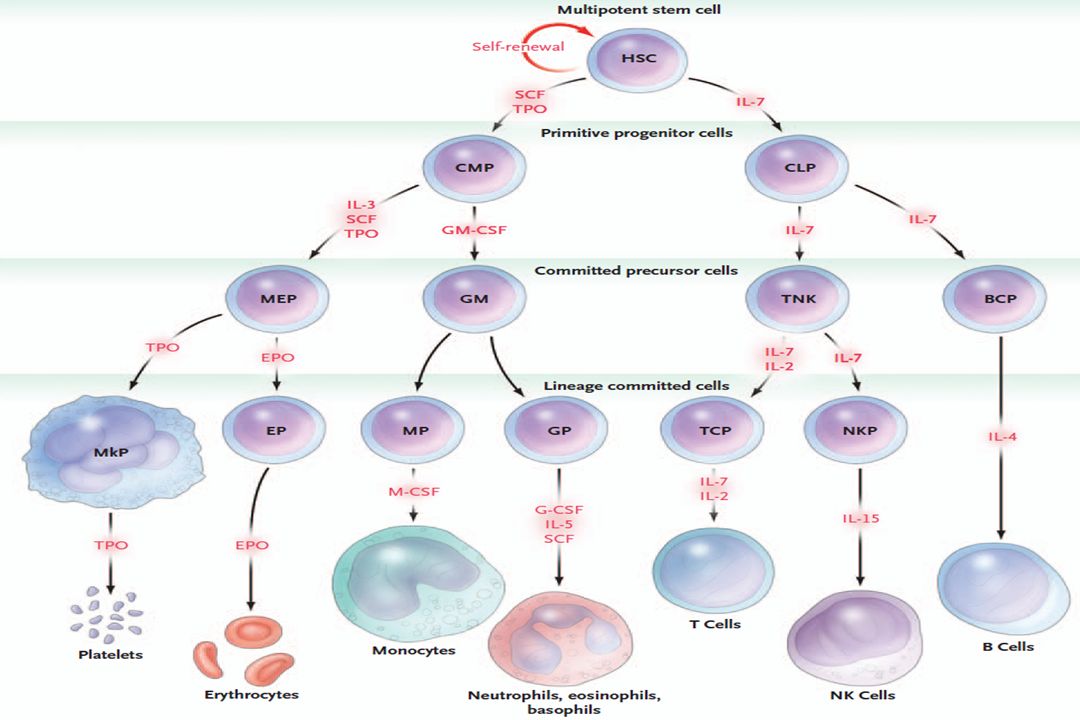
Definizione Le leucemie costituiscono le neoplasie delle cellule staminali ematopoietiche. Sono caratterizzate da sostituzione diffusa del midollo osseo da parte di cellule neoplastiche. Nella maggior parte dei casi le cellule leucemiche diffondono nel sangue periferico dove si osservano in notevole numero. Queste cellule possono inoltre infiltrare il fegato, la milza, i linfonodi ed altri tessuti.

5
Basso grado di maturità
Classificazione Le leucemie vengono classificate in funzione del grado di maturità delle cellule leucemiche stesse. Acute Croniche Basso grado di maturità Alto grado di maturità e del tipo cellulare predominante Mieloide Linfoide

6
Definizione di leucemia acuta
Il termine acuta si riferisce alla rapidità di insorgenza dei sintomi e della progressione della malattia che, se non curata, può condurre a morte in breve tempo, a differenza della leucemia cronica il cui decorso è in genere molto meno tumultuoso. Esso non implica un giudizio negativo poiché molti casi di leucosi acuta possono essere curati con la sola chemioterapia

7
Leucemie Acute Sono malattie neoplastiche caratterizzate dalla proliferazione incontrollata di cellule emopoietiche molto immature nel midollo BLASTI

8
BLASTI Cellule che hanno perso la capacità di dare origine alle cellule più mature che poi passano nel sangue periferico e che continuano a dividersi, aumentando progressivamente di numero Sangue midollare blasti > 25% Sangue periferico blasti >5%

10
Neoplasie del sistema emopoietico e linfoide
Neoplasie linfoidi Le cellule neoplastiche esprimono le caratteristiche di particolari stadi della differenziazione linfocitaria Leucemie linfoidi Linfomi Hodgkin e non Hodgkin Gammopatie monoclonali Neoplasie mieloidi Linea mieloide – eritrocitaria, granulocitaria, monocitaria, megacariocitaria Leucemie mieloidi acute Sindromi mielodisplastiche (MDS) Disordini mieloproliferativi cronici Istiocitosi Rari disordini proliferativi di macrofagi e DC
 Disordini mieloproliferativi cronici. Istiocitosi. Rari disordini proliferativi di macrofagi e DC.")
11
Eziologia Sostanzialmente sconosciute.
Sicuramente non è ereditaria, cioè non può essere trasmessa dai genitori ai figli: può essere familiare. C'è il sospetto, ma non la prova definitiva, che alcune sostanze possano provocare una leucemia linfoblastica acuta: le radiazioni ionizzanti (le leucemie acute sono più frequenti nei sopravvissuti alle bombe atomiche di Hiroshima e Nagasaki o in soggetti trattati con radioterapia per altre neoplasie;) benzene, sostanza contenuta nel petrolio e nella benzina; alcuni farmaci usati per la cura dei tumori (ma non tutti!), specie se usati in combinazione con la radioterapia
 benzene, sostanza contenuta nel petrolio e nella benzina; alcuni farmaci usati per la cura dei tumori (ma non tutti!), specie se usati in combinazione con la radioterapia.")
12
Origine dalla linea linfocitaria
Neoplasie linfoidi Origine dalla linea linfocitaria

13
Linfoma vs leucemia linfocitica
Esteso coinvolgimento tumorale del midollo con presenza di un gran numero di cellule tumorali nel sangue periferico Linfoma Proliferazione cellulare con formazione di una massa discreta La linea di separazione è piuttosto virtuale Molti linfomi possono mostrare la presenza in circolo di cellule tumorali e un esteso coinvolgimento midollare → evoluzione in forma leucemica Molte leucemie originano come masse di tessuto senza coinvolgimento midollare L’utilizzo dei due termini dunque deriva dalla valutazione della differente presentazione della malattia al momento della diagnosi

14
Neoplasie linfoidi Linfomi Hodgkin (HL) e non Hodgkin (NHL)
Forme clinicamente e istologicamente differenti – implicazioni terapeutiche Neoplasie plasmacellulari Tumori costituiti da cellule B differenziate a livello terminale localizzati quasi interamente a livello midollare – raro interessamento dei linfonodi o ‘leucemico’ Secrezione di immunoglobuline → gammopatie monoclonali

15
Presentazione clinica
Linfomi 2/3 dei NHL e quasi la totalità dei HL si presentano come una tumefazione linfonodale di discreta consistenza, spesso > 2 cm Il restante 1/3 dei NHL si presenta in siti extranodali (stomaco, epidermide) Leucemie Le leucemie frequentemente esordiscono con sintomi e segni collegati alla soppressione della ematopoiesi a causa della invasione midollare Le leucemie linfocitiche infiltrano la milza e il fegato Le neoplasie plasmacellulari che interessano lo scheletro causano distruzione locale del tessuto, presentandosi dunque come forme dolorose per fratture patologiche
 Leucemie. Le leucemie frequentemente esordiscono con sintomi e segni collegati alla soppressione della ematopoiesi a causa della invasione midollare. Le leucemie linfocitiche infiltrano la milza e il fegato. Le neoplasie plasmacellulari che interessano lo scheletro causano distruzione locale del tessuto, presentandosi dunque come forme dolorose per fratture patologiche.")
16
REAL Revised European-American Classification of Lymphoid Neoplasms (1994) → classificazione delle neoplasie linfoidi Caratteristiche cliniche, morfologiche Immunofenotipo, alterazioni genetiche La REAL ha mostrato una riproducibilità diagnostica e una capacità di stratificare i pazienti in base alla prognosi soddisfacente Revisione da parte della WHO (1997)
")
17
REAL Neoplasie dei precursori delle cellule B
Cellule B immature Neoplasie delle cellule B periferiche Cellule B mature Neoplasie dei precursori delle cellule T Cellule T immature Neoplasie delle cellule T e NK periferiche Cellule T e NK mature Linfomi Hodgkin Cellule di Reed-Sternberg e varianti

18
Leucemia linfoblastica acuta/ Linfoma linfoblastico acuto (ALL)
85% origine pre-B Restante origine pre-T Neoplasia infantile

19
Neoplasie dei precursori T e B
Leucemia/linfoma linfoblastico acuto (ALL) Linfoblasti (pre-T o pre-B, morfologicamente indistinguibili - immunofenotipizzazione) 85% di origine pre-B → forma leucemica acuta nei bambini, con esteso coinvolgimento midollare e del sangue periferico Meno comunemente di origine pre-T → linfomi in maschi adolescenti, con frequente coinvolgimento timico (50-70%) (possibile evoluzione leucemica)
 Linfoblasti (pre-T o pre-B, morfologicamente indistinguibili - immunofenotipizzazione) 85% di origine pre-B → forma leucemica acuta nei bambini, con esteso coinvolgimento midollare e del sangue periferico. Meno comunemente di origine pre-T → linfomi in maschi adolescenti, con frequente coinvolgimento timico (50-70%) (possibile evoluzione leucemica)")
20
Epidemiologia 2500 nuovi casi / anno USA
Gran parte in bambini < 15 anni Rappresenta la malattia tumorale più frequente nei bambini Più comune nei bianchi e nei maschi Pre-B ALL → picco di incidenza a 4 anni Coincide con il picco del numero di linfoblasti pre-B a livello midollare Pre-T ALL → picco di incidenza in adolescenza Massimo sviluppo timico Entrambe le forme possono manifestarsi ad ogni età, cmq con una frequenza minore che nell’infanzia

21
FAB classification L1 L2 L3 Piccoli blasti omogenei (bambino)
Blasti eterogenei per dimensioni e caratteristiche (adulto) L3 Grandi blasti a citoplasma basofilo ed ipervacuolato
 L3. Grandi blasti a citoplasma basofilo ed ipervacuolato.")
22
Caratteristiche cliniche
ALL molto simile a AML Anemia, neutropenia, trombocitopenia L’accumulo delle cellule neoplastiche immature interferisce con la normale emopoiesi midollare Danno meccanico da occupazione fisica e distorsione dell’architettura midollare Competizione per fattori di crescita e nutrienti Secrezione di fattori di inibizione Infiltrazione in organi e tessuti linfoidi e non (sindrome da massa tumorale) Localizzazione splenica, epatica, linfoadenopatia, CNS, testicoli Liberazione di citochine e mediatori umorali Febbre, dolori ossei e muscolari, stato cachettico, sudorazioni notturne
 Localizzazione splenica, epatica, linfoadenopatia, CNS, testicoli. Liberazione di citochine e mediatori umorali. Febbre, dolori ossei e muscolari, stato cachettico, sudorazioni notturne.")
23
Caratteristiche cliniche
Esordio improvviso e violento Il paziente si presenta dopo pochi giorni o settimane dall’esordio dei sintomi Sintomi correlati all’insufficienza midollare Anemia → fatica, pallore, dispnea Neutropenia → febbre, infezioni Trombocitopenia → petecchie, ecchimosi, epistassi, sanguinamento gastrointestinale Dolore osseo e perdita di durezza ossea Espansione midollare e infiltrazione del subperiostio Linfoadenopatia generalizzata, splenomegalia, epatomegalia Più comuni nelle neoplasie linfoidi rispetto alle mieloidi Nelle forme pre-T a carattere linfomatoso con interessamento timico → compressione dei vasi mediastinici e delle vie aeree Manifestazioni centrali → esame del liquor Mal di testa, vomito, paralisi nervose → diffusione meningea, più comune nelle forme linfoidi

24
Leucemia linfoblastica acuta:
marcata linfoadenopatia cervicale in un bambino di 4 anni. (Cortesemente da Prof. JM Chessells)
")
25
Diagnosi 1 Si potrà sospettare la malattia in seguito a:
Sintomi riferiti da voi e/o anomalie riscontrate durante la visita pallore o emorragie cutanee, epatomegalia splenomegalia, linfoadenomegalia Emocromo con formula: aumento dei globuli bianchi (ma a volte si può avere una diminuzione), spesso con anemia e/o piastrinopenia, variamente associate nel singolo caso; inoltre alla formula leucocitaria potrà esserci una percentuale di cellule immature, blasti.
, spesso con anemia e/o piastrinopenia, variamente associate nel singolo caso; inoltre alla formula leucocitaria potrà esserci una percentuale di cellule immature, blasti.")
26
presso un centro specializzato
Diagnosi 2 presso un centro specializzato Agoaspirato: esame morfologico Biopsia osteomidollare: esame istologico Sui campioni prelevati saranno effettuati studi estremamente sofisticati per stabilire il tipo di leucemia la citochimica, l’immunofenotipo, la citogenetica e la biologia molecolare. Sulla base di questi esami viene eventualmente confermata la diagnosi e calcolato il livello di rischio, cioè la probabilità di andare incontro a recidiva

27
Prognosi e terapia Con la chemoterapia aggressiva > 90% dei bambini vanno incontro a completa remissione e almeno 2/3 possono considerarsi guariti Fattori prognostici negativi Età < 2 anni (translocazione del gene MLL) Sesso maschile Esordio in età adolescenziale o adulta → Bcl-Abl+ Conta blastica periferica > /μl Presenza di mutazioni sfavorevoli: t(9;22)Ph+ nel 3% ALL infantili 30% ALL adulti Malattia minima residua dopo trattamento → rischio di recidiva Fattori prognostici positivi Età 2-10 anni Bassa conta leucocitaria Fenotipo pre-B precoce Iperploidia o t(12;21) Trapianto allogenico di midollo offre speranze nelle categorie a prognosi sfavorevole
 Sesso maschile. Esordio in età adolescenziale o adulta → Bcl-Abl+ Conta blastica periferica > /μl. Presenza di mutazioni sfavorevoli: t(9;22)Ph+ nel 3% ALL infantili 30% ALL adulti. Malattia minima residua dopo trattamento → rischio di recidiva. Fattori prognostici positivi. Età 2-10 anni. Bassa conta leucocitaria. Fenotipo pre-B precoce. Iperploidia o t(12;21) Trapianto allogenico di midollo offre speranze nelle categorie a prognosi sfavorevole.")
28
Terapia Induzione della remissione
Remissione – blasti < 5% nel midollo e assenza di cellule lucemiche nel sangue periferico Vincristina, antracicline, corticosteroidi Protezione del sistema nervoso centrale con trattamento profilattico: iniezione intratecale di metotrexate Terapia post-induzione (consolidamento) prevenzione delle recidive (valutazione del rischio) Trapianto autologo o eterologo (giovani) Chemoterapia Antiblastici, metotrexate, ciclofosfamide, citosina arabinoside Obiettivo → induzione della remissione completa e prevenzione delle recidive
 prevenzione delle recidive (valutazione del rischio) Trapianto autologo o eterologo (giovani) Chemoterapia. Antiblastici, metotrexate, ciclofosfamide, citosina arabinoside. Obiettivo → induzione della remissione completa e prevenzione delle recidive.")
29
10-20% No remissione morte induzione Recidiva precoce 90% bambini 75-90% adulti remissione Nuovo ciclo di induzione consolidamento Trapianto autologo Trapianto eterologo Nessuna terapia Polichemoterapia guarigione morte recidiva

30
(eritrociti, granulociti, monociti e megacariociti)
Neoplasie mieloidi Origine dalle cellule ematopoietiche in grado di differenziare nella linea mieloide (eritrociti, granulociti, monociti e megacariociti)
")
31
Introduzione Coinvolgimento midollare prevalente
Emopoiesi alterata Minore interessamento degli organi emopoietici secondari (milza, fegato, linfonodi)
")
32
Neoplasie mieloidi Leucemie mieloidi acute Sindromi mielodisplastiche
Accumulo nel midollo di blasti immaturi con soppressione della normale attività midollare Sindromi mielodisplastiche Emopoiesi inefficace → citopenia Disordini mieloproliferativi cronici Aumentata produzione di cellule mieloidi terminalmente differenziate

33
Considerazioni Le specifiche manifestazioni delle neoplasie mieloidi dipendono Dalla posizione della cellula colpita nella gerarchia emopoietica Dall’entità della compromissione della differenziazione con blocco di una linea in favore di altre differenziazioni Classificazione in base al corrispettivo morfologico normale

34
Leucemia mieloide acuta
Interessa principalmente i giovani adulti (15-39 anni) ma frequente anche in altre età più avanzate o nei bambini Forme primarie No fattori di rischio dimostrabili Forme secondarie Fattori di rischio come esposizioni a leucemogeni, terapia genotossica, predisposizione genetica (anemia di Fanconi, sindrome di Bloom), MDS 3-10% dei pazienti trattati con terapia alchilante per HL, NHL, tumori ovarici, MM → AML
 ma frequente anche in altre età più avanzate o nei bambini. Forme primarie. No fattori di rischio dimostrabili. Forme secondarie. Fattori di rischio come esposizioni a leucemogeni, terapia genotossica, predisposizione genetica (anemia di Fanconi, sindrome di Bloom), MDS. 3-10% dei pazienti trattati con terapia alchilante per HL, NHL, tumori ovarici, MM → AML.")
35
Fisiopatologia Inibizione della differenziazione
Accumulo di blasti relativamente indifferenziati che soppiantano la normale popolazione midollare Possibilità di uno o più tipi di differenziazione mieloide La velocità di replicazione è più bassa dei normali precursori → importanza della alterazione differenziativa e dell’aumentata sopravvivenza Soppressione della normale attività midollare → citopenia (anemia, trombocitopenia, neutropenia)
")
37
Classificazione FAB classification (French-American-British) → otto categorie (M0 → M7) Grado di differenziazione e orientamento Analisi immunoistochimica per la MPO, esterasi specifiche e non specifiche, markers molecolari WHO classification Categorie in base alle alterazioni citogenetiche ed ai meccanismi patogenetici → utilità terapeutica della classificazione

38
HSC CFU-Blast Precursore timico M0 CFU-GEMM CFU-Eos CFU-B/Mast M1 BFU-EM CFU-GM M2 CFU-MK CFU-G CFU-E CFU-M M6 M7 Mieloblasto Promielocito Mielocito Metamielocito Forme a Banda M3 Monoblasto Promonocito Monocito M5a M4 M5b Monociti (esterasi non-specifiche) Neutrofili (MPO+, Auer-rods)
 Neutrofili. (MPO+, Auer-rods)")
39
FAB classification M0 → AML minimamente differenziata
I blasti mancano di markers citologici (MPO-) di differenziamento mieloide → presenza solo di antigeni della linea mieloide M1 → AML senza differenziazione I blasti (MPO+) sono molto immaturi M2 → AML con maturazione I blasti presentano una tendenza differenziativa granulocitaria M3 → PML (leuc. promielocitica acuta) Gran parte delle cellule sono promielociti ipergranulari M4 → L. monomielocitica (o monoblastica) acuta Evidente differenziamento mielocitico e monocitico (monoblasti) M5 → L. monocitica acuta Differenziamento monoblastico (M5a) o monocitico (M5b) M6 → Eritroleucemia acuta Precursori eritroidi displastici M7 → Leucemia megacariocitica acuta Blasti della linea megacariocitica
 di differenziamento mieloide → presenza solo di antigeni della linea mieloide. M1 → AML senza differenziazione. I blasti (MPO+) sono molto immaturi. M2 → AML con maturazione. I blasti presentano una tendenza differenziativa granulocitaria. M3 → PML (leuc. promielocitica acuta) Gran parte delle cellule sono promielociti ipergranulari. M4 → L. monomielocitica (o monoblastica) acuta. Evidente differenziamento mielocitico e monocitico (monoblasti) M5 → L. monocitica acuta. Differenziamento monoblastico (M5a) o monocitico (M5b) M6 → Eritroleucemia acuta. Precursori eritroidi displastici. M7 → Leucemia megacariocitica acuta. Blasti della linea megacariocitica.")
41
Caratteristiche cliniche
ALL molto simile a AML L’accumulo delle cellule neoplastiche (blasti > 30% nel midollo) immature interferisce con la normale emopoiesi midollare Danno meccanico da occupazione fisica e distorsione dell’architettura midollare Competizione per fattori di crescita e nutrienti Secrezione di fattori di inibizione Anemia, neutropenia, trombocitopenia Infiltrazione di tessuti non emopoietici Liberazione di mediatori solubili (citochine)
 immature interferisce con la normale emopoiesi midollare. Danno meccanico da occupazione fisica e distorsione dell’architettura midollare. Competizione per fattori di crescita e nutrienti. Secrezione di fattori di inibizione. Anemia, neutropenia, trombocitopenia. Infiltrazione di tessuti non emopoietici. Liberazione di mediatori solubili (citochine)")
42
Caratteristiche cliniche
Esordio improvviso e violento Il paziente si presenta dopo pochi giorni o settimane dall’esordio dei sintomi Sintomi correlati all’insufficienza midollare Anemia → fatica, pallore, dispnea Neutropenia → febbre, infezioni opportunistiche Trombocitopenia → petecchie, ecchimosi, epistassi, sanguinamento gastrointestinale DIC (leucemia promielocitica acuta – M3) Leucocitosi (non costante) → segni di leucostasi e leucotrombosi Dolore osseo e perdita di durezza ossea Espansione midollare e infiltrazione del subperiostio Linfoadenopatia generalizzata, splenomegalia, epatomegalia Più comuni nelle neoplasie linfoidi rispetto alle mieloidi Differenziazione monocitica (M4, M5) → infiltrazione cutanea (leucemia cutanea) Manifestazioni centrali (più comuni nella ALL)
 Leucocitosi (non costante) → segni di leucostasi e leucotrombosi. Dolore osseo e perdita di durezza ossea. Espansione midollare e infiltrazione del subperiostio. Linfoadenopatia generalizzata, splenomegalia, epatomegalia. Più comuni nelle neoplasie linfoidi rispetto alle mieloidi. Differenziazione monocitica (M4, M5) → infiltrazione cutanea (leucemia cutanea) Manifestazioni centrali (più comuni nella ALL)")
43
Leucostasi e Leucotrombosi

44
infezioni, causa della mancanza di globuli bianchi
Leucemia mieloide acuta: è stato isolato Staphylococcus aureus (a) da un'infezione dell’orbita destra e tessuto circostante
 da un infezione dell’orbita destra e tessuto circostante.")
45
Leucemia mieloide acuta: infezione da aspergillo
scansione TC che mostra una lesione capsulata del cervello nella zona frontale destra, causata da infezione da aspergillo circondata da un'area ipodensa causata dall'infiammazione

46
↓Piastrine petecchie ecchimosi epistassi ematomi

47
Leucemia mieloide acuta:
petecchie emorragiche che ricoprono la parte superiore del torace e la faccia in una piastrinopenia grave.

48
Leucemia mieloide acuta:
ecchimosi marcate, petecchie emorragiche e lividi sull’inguine e sulla coscia;

49
Caratteristiche particolari
Leucemia promielocitica acuta (M3) Promielociti ipergranulati RARα-PML t(15;17) → sensibilità all’acido retinoico Alta incidenza di DIC → fenomeni emorragici Associazione retinoidi / antracicline Leucemia monoblastica acuta (M4) Infiltrazione della cute (leukemia cutis) e delle mucose (gengive) Elevate quantità di lisozima nel plasma Leucemia megacariocitica acuta (M7) Anticorpi contro gpIIb/IIIa, vWF
 Promielociti ipergranulati. RARα-PML t(15;17) → sensibilità all’acido retinoico. Alta incidenza di DIC → fenomeni emorragici. Associazione retinoidi / antracicline. Leucemia monoblastica acuta (M4) Infiltrazione della cute (leukemia cutis) e delle mucose (gengive) Elevate quantità di lisozima nel plasma. Leucemia megacariocitica acuta (M7) Anticorpi contro gpIIb/IIIa, vWF.")
50
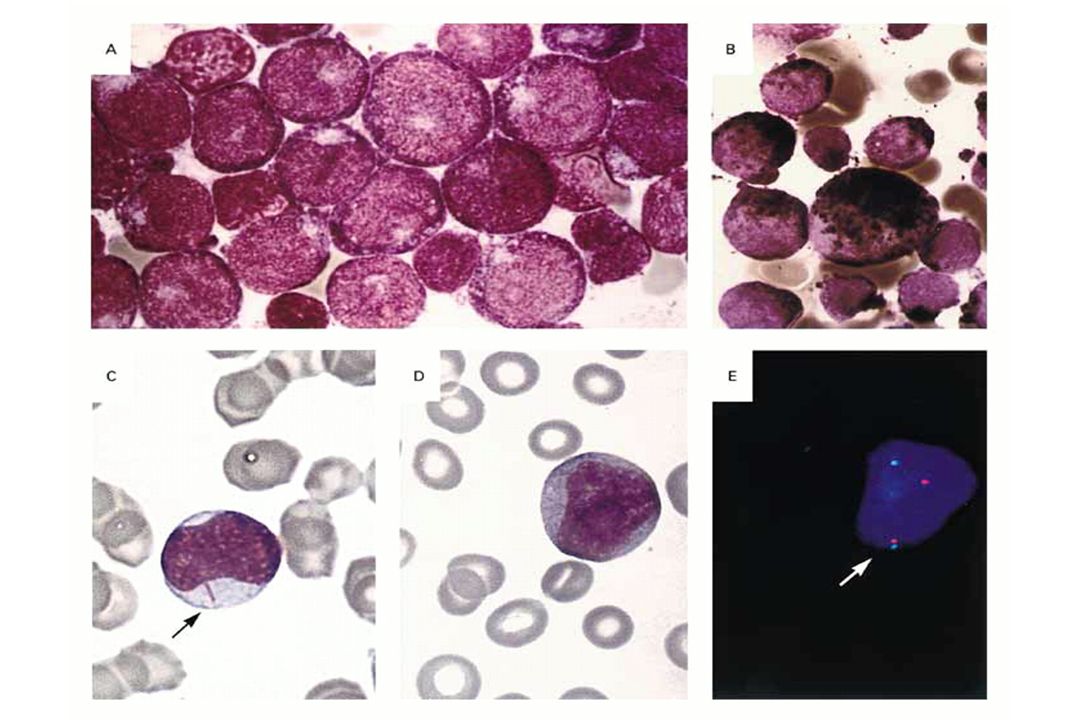
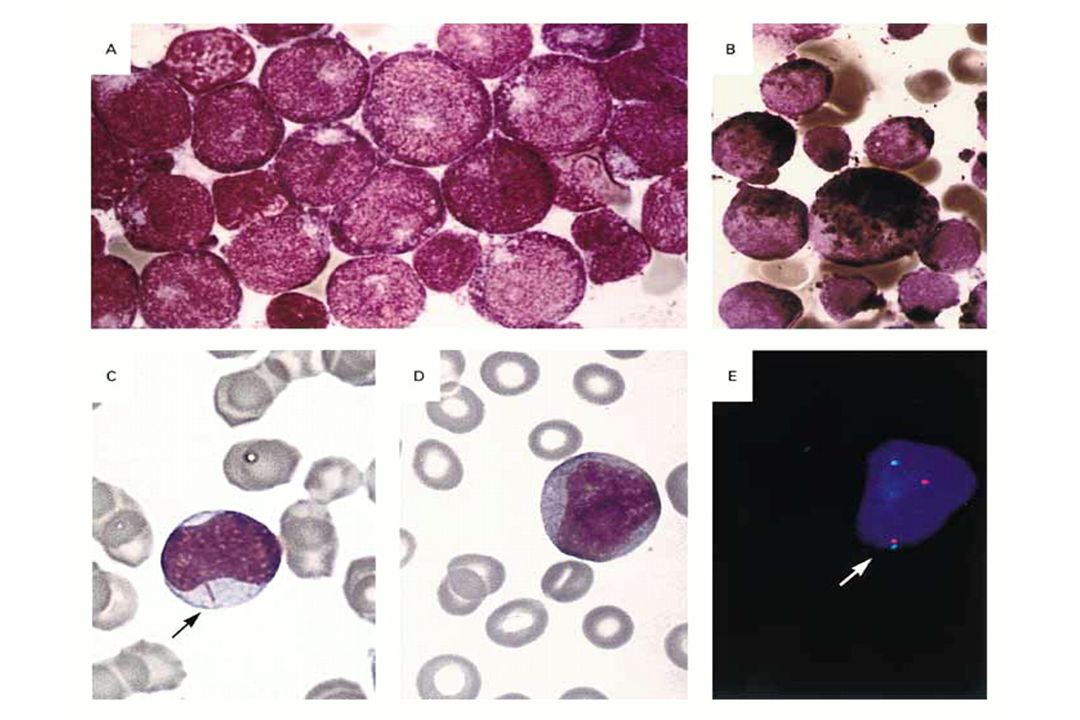
Leucemia Acuta Promielocitica (M3 FAB)
")
51
Leukemic Gingival Infiltration
Leukemic infiltration of the gingivae has been associated with monocytic variants of AML (M4) Mani A, N Engl J Med 2008;258:274
 Mani A, N Engl J Med 2008;258:274.")
52
Gruppi prognostici Prognosi favorevole (20%) (frequente la completa guarigione (>85%) e scarso rischio di ricaduta (30-40%) Età <60 anni t(15;17), t(8;21), inv(16) Prognosi intermedia No alterazioni citogenetiche Prognosi sfavorevole (15%) Età avanzata o forme secondarie Alterazioni citogenetiche multiple
, t(8;21), inv(16) Prognosi intermedia. No alterazioni citogenetiche. Prognosi sfavorevole (15%) Età avanzata o forme secondarie. Alterazioni citogenetiche multiple.")
53
Terapia Induzione della remissione
Remissione – blasti < 5% nel midollo e assenza di cellule lucemiche nel sangue periferico Daunorubicina e citarabina 70-80% dei pazienti <60 anni e 50% nei pazienti più anziani Terapia post-induzione (consolidamento) prevenzione delle recidive (valutazione del rischio) Trapianto autologo o eterologo (giovani) Chemoterapia Obiettivo → induzione della remissione completa e prevenzione delle recidive
 prevenzione delle recidive (valutazione del rischio) Trapianto autologo o eterologo (giovani) Chemoterapia. Obiettivo → induzione della remissione completa e prevenzione delle recidive.")
54
Trapianto eterologo allogenico
Terapia mieloablativa sovramassimale + trapianto di cellule staminali da donatore HLA compatibile Azione mieloablativa Graft versus leukemia effect Complicanze Immunosoppressione → infezioni, neoplasie Graft versus host disease Mortalità associata al trapianto → 5-15% Indicazioni → alto rischio di recidiva in pazienti < 55 anni Scarso rischio di recidive

55
Trapianto autologo Minori complicanze collegate al tumore
Assenza di GVHD Aumentato rischio di recidive Assenza di GVL effect Possibile ‘contaminazione’ delle cellule staminali reinfuse

56
Forme secondarie Terapia citotossica alchilante (HL, NHL, tumori ovarici e MM) MDS → AML, esordio a 5-10 anni Terapia con inibitori della topoisomerasi II (epipodofillotossina) AML non preceduta da MDS, esordio precoce Frequente associazione con alterazioni di MLL (11q23)
 AML non preceduta da MDS, esordio precoce. Frequente associazione con alterazioni di MLL (11q23)")
57
Terapia di Supporto Trasfusione di Globuli rossi (per migliorare l’anemia), Piastrine (per prevenire o curare emorragie), Antibiotici, antifungini e antivirali somministrati per prevenire o curare infezioni in atto; Fattori di crescita, farmaci somministrati per ridurre la leucopenia; la nutrizione parenterale in caso di impossibilità a nutrirsi spontaneamente, spesso a causa delle complicanze della chemioterapia e della radioterapia; la terapia antidolorfica; il supporto psicologico ecc. La terapia di supporto consente quindi consente di attenuare o prevenire alcuni degli effetti collaterali più gravi, rendendo possibile la continuazione a cicli periodici della terapia vera e propria.

58
Leucemia Linfocitica Cronica (CLL) - Linfoma a piccoli linfociti (SLL)
Linfociti B maturi Rappresenta la forma leucemica più comune nella razza bianca (25%) nuovi casi anno USA Età media 60 anni Prognosi altamente variabile
 nuovi casi anno USA. Età media 60 anni. Prognosi altamente variabile.")
59
Leucemia linfocitica cronica (CLL) / linfoma a piccoli linfociti (SLL)
Origine dalle cellule B mature (linfociti) Le due forme sono indistinguibili da un punto di vista morfologico, fenotipico e genetico → la differenza è nell’entità dei linfociti neoplastici nel sangue periferico Linfociti > 4000/µl → leucemia La CLL è la leucemia più comune nei paesi occidentali SLL → 4% dei NHL
 Le due forme sono indistinguibili da un punto di vista morfologico, fenotipico e genetico → la differenza è nell’entità dei linfociti neoplastici nel sangue periferico. Linfociti > 4000/µl → leucemia. La CLL è la leucemia più comune nei paesi occidentali. SLL → 4% dei NHL.")
60
Caratteristiche cliniche
Il paziente è spesso asintomatico Età > 50 anni (60 in media) ♂/♀ = 2/1 Sintomi aspecifici – affaticabilità, anoressia, calo ponderale Linfoadenopatia ed epatosplenomegalia Indolenti, non duri, mobili sui piani superficiali e profondi, no tendenza a confluire in pacchetti o fistolizzare Leucopenia (SLL) o forte leucocitosi (> /μl) Anemia immunoemolitica o trombocitopenia (10-15%) La malattià ha un decorso molto variabile → difficoltà nel prevedere l’evoluzione Sistema di stadiazione di Rai e Binet (clinica) Valutazione della ipermutazione IgVh
 ♂/♀ = 2/1. Sintomi aspecifici – affaticabilità, anoressia, calo ponderale. Linfoadenopatia ed epatosplenomegalia. Indolenti, non duri, mobili sui piani superficiali e profondi, no tendenza a confluire in pacchetti o fistolizzare. Leucopenia (SLL) o forte leucocitosi (> /μl) Anemia immunoemolitica o trombocitopenia (10-15%) La malattià ha un decorso molto variabile → difficoltà nel prevedere l’evoluzione. Sistema di stadiazione di Rai e Binet (clinica) Valutazione della ipermutazione IgVh.")
61
Linfoadenopatia nella CLL/SLL

62
Stadiazione secondo Rai e Binet
Basso rischio (interessati < 3 siti linfonodali) Stadio 0 Linfociti nel sangue >15.000/μl e midollo > 40% Rischio intermedio (interessati > 3 siti linfonodali) Stadio 1 Come stadio 0 + linfoadenomegalia Stadio 2 Come stadio 0 + epatosplenomegalia, con o senza linfoadenomegalia Alto rischio (anemia e/o piastrinopenia) Stadio 3 Come stadio 0 + anemia, con o senza interessamento epatosplenico o linfonodale Stadio 4 Come stadio 0 + piastrinopenia, con o senza anemia, interessamento epatosplenico o linfonodale
 Stadio 0. Linfociti nel sangue >15.000/μl e midollo > 40% Rischio intermedio (interessati > 3 siti linfonodali) Stadio 1. Come stadio 0 + linfoadenomegalia. Stadio 2. Come stadio 0 + epatosplenomegalia, con o senza linfoadenomegalia. Alto rischio (anemia e/o piastrinopenia) Stadio 3. Come stadio 0 + anemia, con o senza interessamento epatosplenico o linfonodale. Stadio 4. Come stadio 0 + piastrinopenia, con o senza anemia, interessamento epatosplenico o linfonodale.")
63
Evoluzione Trasformazione prolinfocitica (15-30%)
Citopenia, splenomegalia, prolinfociti nel sangue periferico (grande nucleo con nucleolo centrale prominente) Sindrome di Richter (10%) → linfoma a grandi cellule B diffuso Massa a rapida crescita nella milza o linfonodi Prognosi molto severa (sopravvivenza < 1 anno) Infezioni, anemia emolitica autoimmune, aplasia midollare
 Sindrome di Richter (10%) → linfoma a grandi cellule B diffuso. Massa a rapida crescita nella milza o linfonodi. Prognosi molto severa (sopravvivenza < 1 anno) Infezioni, anemia emolitica autoimmune, aplasia midollare.")
64
Leucemia prolinfocitica acuta
Insorgenza de novo o da un quadro di CLL Maschio adulto-anziano (60-65 anni) Accumulo di cellule linfoidi (prolinfociti) con ampio citoplasma, nucleo eucromatico con nucleolo rilevante, tipologia B Invasione midollare (citopenia) e degli organi linfoidi (milza, fegato, linfonodi) Leucocitosi spesso molto marcata (prolinfociti) Splenoepatomegalia
 Accumulo di cellule linfoidi (prolinfociti) con ampio citoplasma, nucleo eucromatico con nucleolo rilevante, tipologia B. Invasione midollare (citopenia) e degli organi linfoidi (milza, fegato, linfonodi) Leucocitosi spesso molto marcata (prolinfociti) Splenoepatomegalia.")
65
Terapia Pazienti asintomatici, soprattutto in età avanzata → no terapia Contenere la malattia Terapia aggressiva se vi sono evoluzioni Pazienti giovani, forme aggressive → terapia citotossica aggressiva con indicazione al trapianto eterologo

Presentazioni simili




.>")
.>")




>")








