Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
FISIOLOGIA DELL’ACCRESCIMENTO
Corso di Pediatria Riconversione crediti Fisioterapia Cattedra di Pediatria Facoltà di Medicina e Chirurgia Università degli Studi di Chieti FISIOLOGIA DELL’ACCRESCIMENTO

2
La crescita Il soggetto in età evolutiva aumenta le proprie dimensioni (accrescimento) e contemporaneamente modifica, in modo progressivo, forma e composizione corporea (maturazione o sviluppo). La monitorizzazione dei processi di accrescimento e sviluppo rappresenta un indicatore ottimale per la valutazione delle condizioni di salute individuali, sia per la sorveglianza epidemiologica a livello di popolazione.
 e contemporaneamente modifica, in modo progressivo, forma e composizione corporea (maturazione o sviluppo). La monitorizzazione dei processi di accrescimento e sviluppo rappresenta un indicatore ottimale per la valutazione delle condizioni di salute individuali, sia per la sorveglianza epidemiologica a livello di popolazione.")
3
Formule per calcolare l’altezza media
Alla nascita cm A 1 anno cm 2-12 anni età (anni) x
 x")
4
Velocità di crescita (cm/anno)
Alla nascita : 50cm 1° anno: + 25 cm (incremento della statura pari al 50% della lunghezza neonatale) 75 cm 2° anno: cm (incremento della statura pari al 50% della crescita nel 1° anno) 87.5 cm 3° anno: + 8 cm 95.5 cm 4° anno: + 7 cm (nel quarto anno, raddoppio lunghezza neontatale) ~ 100 cm 5°-10° anno: + 5 cm 10°-18° anno: + 6 – 7 cm incremento staturale di ~ 20 cm (F) con arresto ai 16 anni (picco di velocità di crescita pari a 8,5 cm/aa ad 11.5 anni con rallentamento fino ad arrestarsi ai 16 anni) incremento staturale di ~ 30 cm (M) con arresto ai 18 anni (picco di velocità di crescita pari a 9.5 cm/aa a aa con rallentamento fino ad arrestarsi ai 18 anni)
 75 cm. 2° anno: cm (incremento della statura pari al 50% della crescita nel 1° anno) 87.5 cm. 3° anno: + 8 cm 95.5 cm. 4° anno: + 7 cm (nel quarto anno, raddoppio lunghezza neontatale) ~ 100 cm. 5°-10° anno: + 5 cm. 10°-18° anno: + 6 – 7 cm. incremento staturale di ~ 20 cm (F) con arresto ai 16 anni (picco di velocità di crescita pari a 8,5 cm/aa ad 11.5 anni con rallentamento fino ad arrestarsi ai. 16 anni) incremento staturale di ~ 30 cm (M) con arresto ai 18 anni (picco di velocità di. crescita pari a 9.5 cm/aa a 13.5 aa con rallentamento fino ad arrestarsi ai 18. anni)")
5
Formule per calcolare il peso medio
Alla nascita 3.25 Kg 3-12 mesi età (mesi) + 9 / anni età (anni) x anni età (anni) x / 2
 + 9 / anni età (anni) x anni età (anni) x / 2")
6
Formule per il calcolo del peso medio
Primi 2 anni: velocità di crescita ponderale elevata Nascita ~ 3 Kg Peso raddoppia a 5 mesi ~ 6 Kg (x 2) Peso triplica a 1 anno ~ 9 Kg (x 3) Peso quadruplica a 2 anni ~ 12 Kg (x 4) Dai 2 ai 5 anni: aumento di 2 Kg/anno Dai 6 ai 10 anni: aumento di 1-2 Kg/anno Dai 10 ai 18 anni: aumento di ~ 20 (23-27) Kg (F) (con picco di incremento a 12 anni) aumento di ~ 30 (27-37) Kg (M) (con picco di incremento a 14 anni) L’aumento del peso alla pubertà segue quello dell’altezza con ritardo di ~5mesi Maschi (Kg /anno) Femmine (Kg /anno) ° anno ° anno ° anno ° anno ° anno ° anno ° anno ° anno
 - Peso triplica a 1 anno ~ 9 Kg (x 3) - Peso quadruplica a 2 anni ~ 12 Kg (x 4) Dai 2 ai 5 anni: aumento di 2 Kg/anno Dai 6 ai 10 anni: aumento di 1-2 Kg/anno Dai 10 ai 18 anni: aumento di ~ 20 (23-27) Kg (F) (con picco di incremento a 12 anni) aumento di ~ 30 (27-37) Kg (M) (con picco di incremento a 14 anni) L’aumento del peso alla pubertà segue quello dell’altezza con ritardo di. ~5mesi. Maschi (Kg /anno) Femmine (Kg /anno) 11° anno ° anno ° anno ° anno ° anno ° anno ° anno ° anno")
7
Circonferenza cranica
Femmine 0-36 mesi altezza e peso Circonferenza cranica Maschi CDC, 2000

8
Femmine 2-12 anni altezza e peso BMI Maschi CDC, 2000

9
Centili Italiani di riferimento
Cacciari E et al, Eur J Clin Nutr 2002

10
Centili dell’Italia Centro-Settentrionale
Cacciari E et al, Eur J Clin Nutr 2002

11
Centili dell’Italia Meridionale
Cacciari E et al, Eur J Clin Nutr 2002

12
Curve speciali Sindrome di Turner

13
Sindrome di Williams: curve di crescita

14
Curve speciali: sindrome di Down (girls)
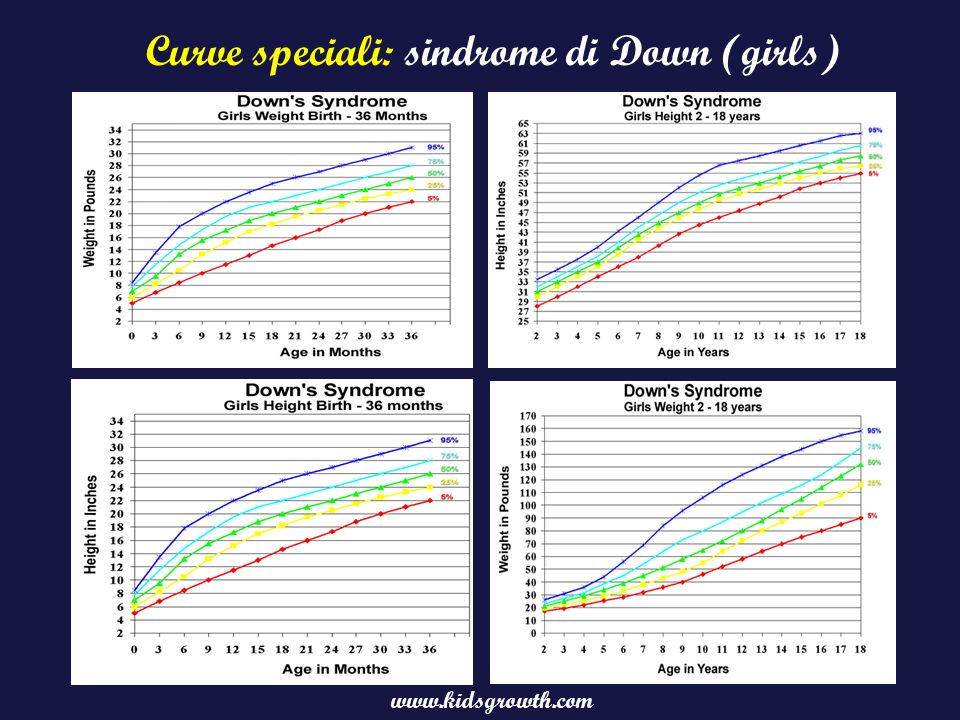
15
Curve speciali: Sindrome di Down (boys)

16
Executive summar of the Low Birghtweight Workshop, 1994
Curve speciali: Basso peso alla nascita Executive summar of the Low Birghtweight Workshop, 1994

17
Executive summar of the Low Birghtweight Workshop, 1994
Curve speciali: Basso peso alla nascita Executive summar of the Low Birghtweight Workshop, 1994

18
Accrescimento patologico
BASSA STATURA: soggetto al di sotto del 3° percentile per età e sesso ALTA STATURA: soggetto al di sopra del 97° percentile per età e sesso VELOCITA’ DI CRESCITA: si misura in cm/anno; occorre un periodo di osservazione di almeno 6 mesi. È patologica se <10° o > 90° percentile

19
Velocità di crescita Tanner JM, Whitehouse RH. Atlas of Children’s Growth

20
Approccio diagnostico
Aspetto fisico (armonico/disarmonico) Accrescimento (percentili, velocità di crescita) Età cronologica Età staturale (età corrispondente al 50° centile per l’ altezza) Target zone Età ossea
 Accrescimento (percentili, velocità di crescita) Età cronologica. Età staturale (età corrispondente al 50° centile per l’ altezza) Target zone. Età ossea.")
21
Target zone Per i maschi: statura padre + statura madre + 13 2
Per le femmine: statura padre + statura madre - 13 ± 5 cm ± 5 cm dove 13 è approssimativamente la differenza fra le stature medie dei maschi e delle femmine

22
Metodo di Greulich e Pyle
Maschio, 8 anni Maschio, 14 anni

23
Anamnesi auxologica Anamnesi familiare Anamnesi prenatale e perinatale
Anamnesi neonatale Anamnesi fisiologica Anamnesi patologica remota Anamnesi patologica prossima

24
Visita auxologica Aspetto fisico, eventuali aspetti dismorfici
Rilevazione delle misure: peso statura statura da seduto circonferenza cranica composizione corporea Rilevazione dello stato puberale Rilevazione del grado di maturazione biologica Standard di crescita Target zone

25
LA PUBERTA’ La pubertà rappresenta quel periodo di transizione tra l’età infantile e l’età adulta, durante il quale avviene lo scatto di crescita, compaiono i caratteri sessuali secondari, si raggiunge la fertilità e avvengono profondi mutamenti psicologici. Pinyerd B et al., J Pediatr Nurs 2005;20:75-82

26
Ojeda & Heger, J Ped Endocrinol Metab 2001

27
LH/FSH fetus infancy childhood puberty adult

28
VALUTAZIONE DELLO SVILUPPO PUBERALE
MAMMELLE PELI PUBICI P Età prepuberale B 1 Età prepuberale P 2 Età ( ) B 2 Età ( ) B 3 Età ( ) P 3 Età ( ) P 4 Età ( ) B 4 Età ( ) B 5 Età ( ) P 5 Età ( ) Marshall WA, Tanner JM, Arch Dis Child 1948, 44:
 B 2. Età ( ) B 3. Età ( ) P 3. Età ( ) P 4. Età ( ) B 4. Età 13.1 ( ) B 5. Età ( ) P 5. Età ( ) Marshall WA, Tanner JM, Arch Dis Child 1948, 44:")
29
VALUTAZIONE DELLO SVILUPPO PUBERALE
GENITALI PELI PUBICI G 1 Età prepuberale: pene e scroto infantili P Età prepuberale: assenza di peli pubici G 2 Età ( ) P 2 Età ( ) G 3 Età ( ) P 3 Età ( ) P 4 Età ( ) G 4 Età ( ) G 5 Età ( ) P 5 Età ( ) Marshall WA and Tanner JM, Arch Dis Child 1970, 45:13-23
 P 2. Età ( ) G 3. Età ( ) P 3. Età ( ) P 4. Età ( ) G 4. Età 13.7 ( ) G 5. Età ( ) P 5. Età ( ) Marshall WA and Tanner JM, Arch Dis Child 1970, 45:")
31
Velocità di crescita Tanner JM, Whitehouse RH. Atlas of Children’s Growth

32
BASSA STATURA Corso di Pediatria Riconversione crediti Fisioterapia
Cattedra di Pediatria Facoltà di Medicina e Chirurgia Università degli Studi di Chieti BASSA STATURA

33
Bassa statura Si definisce di bassa statura un soggetto la cui altezza si colloca al di sotto del 3° percentile di una popolazione di riferimento dello stesso sesso ed età Più precisamente si dovrebbe parlare di vera ipostaturalità quando il soggetto in esame si colloca tra le -2,5 D.S. e le -3 D.S. dalla mediana Dal punto di vista clinico la definizione di scarso accrescimento si riferisce ad una velocità di crescita inferiore al 25° percentile

34
CAUSE DI BASSA STATURA MALATTIE ENDOCRINE DISPLASIE OSSEE
ANOMALIE CROMOSOMICHE E GENICHE RITARDO DI CRESCITA INTRAUTERINO MALATTIE CRONICHE SISTEMICHE BASSA STATURA PSICOSOCIALE VARIANTI NORMALI DI CRESCITA (ritardo costituzionale di crescita, bassa statura familiare)
")
35
DEFICIT DI ORMONE DELLA CRESCITA (GROWTH HORMONE, GH)
Corso di Pediatria Riconversione crediti Fisioterapia Cattedra di Pediatria Facoltà di Medicina e Chirurgia Università degli Studi di Chieti DEFICIT DI ORMONE DELLA CRESCITA (GROWTH HORMONE, GH)
")
36
Asse Ipotalamo/GH/IGF-1
Neurotrasmettitori, glucosio, amminoacidi, lipidi, temperatura, stress, sonno, esercizio IPOTALAMO GHRH SS IPOFISI GH FEGATO IGF-1 Muscolo Fibroblasti Osso

37
Asse GH-IGF-1 Ipotalamo GHRH Recettore GHRH Ipofisi GH Recettore GH
Tessuti periferici Recettore GH Recettore IGF-1 Qualsiasi disordine, secondario a difetti genetici o acquisiti, a carico della secrezione o dell’azione del GH è responsabile di un fenotipo patologico caratterizzato da BASSA STATURA di tipo ARMONICO

38
Fattori che influenzano la secrezione di GH
STIMOLATORI Fisiologici Sonno Esercizio fisico Stress Iperaminoacidemia Postprandiale Patologici Digiuno Anoressia nervosa Produzione ectopica di GHRH Insufficienza renale cronica Diabete INIBITORI Iperglicemia postprandiale Aumento dei NEFA Obesità Ipotiroidismo Ipertiroidismo Ipercortisolismo

39
Fattori farmacologici
STIMOLATORI Ipoglicemia GHRH aMSH TRH Vasopressina Estrogeni Glucagone Agonisti alfa adrenergici Antagonisti beta adrenergici Precursori della serotonina Agonisti dopaminergici Agonisti GABAergici Agonisti colinergici INIBITORI Somatostatina IGF-1 GH CRH Progesterone Glucocorticoidi Agonisti beta adrenergici Serotonino antagonisti Antagonisti dopaminergici Antagonsitio colinergici

40
Concentrazione plasmatica di IGF-1 in rapporto all’età
0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 5 10 15 20 25 30 35 40 45 50 IGF-1 (U/ml) Concentrazione plasmatica di IGF-1 in rapporto all’età Età (anni)
 Concentrazione plasmatica di IGF-1 in rapporto all’età. Età (anni)")
41
Test da stimolo della secrezione di GH
Effetti collaterali Stimolo Dose Picco GH (min) Insulina (ev) U/kg ipoglicemia Clonidina (os) mg/m ipotensione Arginina (ev) g/kg ipoglicemia Glucagone (im) mg/kg ipoglicemia GHRH (ev) or 2 mcg flushes L-Dopa (os) mg nausea
 Insulina (ev) U/kg ipoglicemia. Clonidina (os) 0.15 mg/m ipotensione. Arginina (ev) 0.5 g/kg ipoglicemia. Glucagone (im) 0.03 mg/kg ipoglicemia. GHRH (ev) 1 or 2 mcg flushes. L-Dopa (os) mg nausea.")
42
Test da stimolo della secrezione di GH
Picco inferiore a 10 ng/ml in due test farmacologici deficit ipofisario

43
Picco inferiore a 3 ng/ml
In caso di dubbi al test da stimolo è utile la determinazione della secrezione integrata del GH fisiologicamente prodotto nel corso di 12 ore notturne (1 prelievo circa ogni 20 minuti) (v.n. superiore a 3 ng/ml) Picco inferiore a 3 ng/ml deficit ipotalamico
 (v.n. superiore a 3 ng/ml) Picco inferiore a 3 ng/ml. deficit ipotalamico.")
44
Test di generazione somatomedinica
Somministrare per 4 giorni 0,1/UI/kg di GH valutando i livelli di IGF-1 al tempo 0 e 12 ore dopo dall’ultima somministrazione Si considera positivo un incremento di GH di almeno 3-4 volte rispetto al basale Se la risposta all’IGF-1 è estremamente ridotta o assente va presa in considerazione la diagnosi di resistenza al GH

45
Difetti del recettore del GHRH

46
Nanismo di Sindh 5 famiglie con numerosi consanguinei
17 (14 M, 3 F) soggetti gravemente affetti Statura media uomini: cm ( , in media SDS) Statura media delle donne: cm ( , in media -8.4 SDS) In prima linea: due maschi affetti di 13 e 12 anni; al centro: 4 uomini adulti affetti (20-27 aa); dietro: 3 adulti non affetti provenienti dalla stessa regione Maheshwari et al, Acta Paediatr Suppl 1997
 soggetti gravemente affetti. Statura media uomini: cm ( , in media -7.8 SDS) Statura media delle donne: cm ( , in media -8.4 SDS) In prima linea: due maschi affetti di 13 e 12 anni; al centro: 4 uomini adulti affetti (20-27 aa); dietro: 3 adulti non affetti provenienti dalla stessa regione. Maheshwari et al, Acta Paediatr Suppl")
47
Difetti del recettore del GHRH
Maheshwari et al, JCEM 1998

48
Difetti del recettore del GHRH
Test da stimolo GH della secrezione di GH GH (ng/ml) picco PB GHRH L-Dopa Clonidina IGF-1 generation test IGF-1 (ng/ml) Basale 0 + 4 > 1000 Maheshwari et al, JCEM 1998
 picco PB. GHRH L-Dopa Clonidina IGF-1 generation test. IGF-1 (ng/ml) Basale > Maheshwari et al, JCEM")
49
Difetti del recettore del GHRH
Stop extracellulare Membrana cellulare citoplasma Maheshwari et al., JCEM 1998 Maheshwari et al, Acta Paediatr Suppl 1997

50
Il difetto di GHRH-R non impedisce la fertilità
statura della madre: 114 cm statura del padre:134 cm Lunghezza alla nascita: 44 cm (-2.9 SDS) Peso alla nascita: 2 kg (-3.4 SDS) Maheshwari et al, Acta Paediatr Suppl 1997
 Peso alla nascita: 2 kg (-3.4 SDS) Maheshwari et al, Acta Paediatr Suppl")
51
Alterazioni dell’asse GH-IGF-1
GHRH IGF-1 Recettore GHRH Ipotalamo Ipofisi Tessuti periferici Recettore GH Recettore IGF-1 GH

52
Difetti del gene del GH Test da stimolo del GH IGF-1 generation test
97 Bambine 50 GH (ng/ml) PB picco clonidina insulina 3 Altezza (cm) IGF-1 generation test GH 0.5 U/kg/settimana IGF-1 (ng/ml) basale +4 40 197 Età (anni)
 PB picco. clonidina insulina Altezza (cm) IGF-1 generation test. GH 0.5 U/kg/settimana. IGF-1 (ng/ml) basale Età (anni)")
53
Classificazione dei deficit di GH di origine genetica
DEFICIT ISOLATO DI GH Tipo Trasmissione GH endogeno Associazioni risposta alla Eziologia terapia con GH GHRH ? Rec. GHRH ? basso nessuna risposta buona mutazioni al GHRH GHRH-R Mutazioni di GH-1 (GHD): IA A indosabile anticorpi anti delezioni e hGH e resistenza mutazioni GH-1 IB AR assente o basso buona mutazioni GH-1 II AD basso iperinsulinemia buona mutazioni GH-1 III X-linked basso ipoglobulinemia Sizonenko PC et al, Growth Hormone& IGF Research 2001
: IA A indosabile - anticorpi anti delezioni e hGH e resistenza mutazioni GH-1. IB AR assente o basso - buona mutazioni GH-1. II AD basso iperinsulinemia buona mutazioni GH-1. III X-linked basso ipoglobulinemia. Sizonenko PC et al, Growth Hormone& IGF Research")
54
Classificazione dei deficit di GH di origine genetica (2)
DEFICIT DI GH COMBINATO Tipo Trasmissione Deficit Eziologia I AR GH, TSH, ACTH, Mutazioni Prop-1 FSH, LH, PRL HESX-1/Hesx-1 II AR GH, TSH, PRL Mutazioni Pit-1 III X-linked GH, TSH, ACTH, ? FSH, LH, Sizonenko PC et al, Growth Hormone& IGF Research 2001

55
GH biologicamente inattivo
È stato descritto un ulteriore tipo di difetto genetico a carico del gene del GH che causa inattività biologica dell’ormone senza alterarne la produzione né le caratteristiche immunologiche. La principale caratteristica dei pazienti affetti da questo tipo di deficit sarà un quadro clinico caratteristico per deficit di GH in presenza di normale risposta ai test da stimolo. Il primo paziente è stato descritto da Takahashi e colleghi: si trattava di un bambino con bassa statura armonica, naso a sella e fronte prominente, nato IUGR (PAN= 2250 g, LAN= 39 cm, EG 41 sett.)
")
56
GH bioinattivo: caso clinico
baseline Età decimale Altezza (cm), SDS , -6.1 Velocità di crescita (cm/aa) Età ossea (anni) GH sierico (ng/ml) basale dopo insulina IGF-1 sierico (ng/ml) (v.n ) IGF-1 generation test (ng/ml) IGFBP-3 (g/ml) (v.n. 2.4 0.8) Dopo GH (0.18 mg/kg/sett.) 6.0 200 ng Il grave deficit di crescita (SDS 6 volte al di sotto della media per l’età e sesso) con bassa velocità di crescita, età ossea ritardata, la normale risposta al test con bassi livelli di IGF-1 e IGFBP-3. Dopo il trattamento con GH si noti l’aumento della velocità di crescita Takahashi Y et al, NEJM 1996
, SDS 84.8, Velocità di crescita (cm/aa) 3.9. Età ossea (anni) 2. GH sierico (ng/ml) basale 7.0 dopo insulina IGF-1 sierico (ng/ml) 38 (v.n ) IGF-1 generation test (ng/ml) 38. IGFBP-3 (g/ml) 1.6 (v.n. 2.4 0.8) Dopo GH (0.18 mg/kg/sett.) ng. Il grave deficit di crescita (SDS 6 volte al di sotto della media per l’età e sesso) con bassa velocità di crescita, età ossea ritardata, la normale risposta al test con bassi livelli di IGF-1 e IGFBP-3. Dopo il trattamento con GH si noti l’aumento della velocità di crescita. Takahashi Y et al, NEJM")
57
Bioinattività del GH Altezza (cm) Test da stimolo del GH
bambine Test da stimolo del GH 97 GH (ng/ml) PB picco insulina 11,0 26,0 glucagone 19,0 41,0 GHRH 3,7 51,0 50 3 Altezza (cm) IGF-1 generation test IGF-1 (U/ml) basale +4 0,22 1,21 GH 8 U/sett Età (anni) Takahashi Y et al, J Clin Invest, 1997
 PB picco. insulina 11,0 26,0. glucagone 19,0 41,0. GHRH 3,7 51, Altezza (cm) IGF-1 generation test. IGF-1 (U/ml) basale +4. 0,22 1,21. GH 8 U/sett. Età (anni) Takahashi Y et al, J Clin Invest,")
58
GH bioinattivo In questo paziente è stata mappata una mutazione sul codone 77 del gene GH -1 dove un’arginina è sostituita con una cisteina Questa mutazione causa una proteina con attività biologica inferiore a al GH naturale e non in grado di stimolare la fosforilazione della tirosina del recettore del GH Gli stessi autori hanno dimostrato una seconda mutazione nell’ esone 4 del gene GH -1 dove un’acido aspartico è sostituito con una glicina in posizione 112 che causa inattività biologica del GH La descrizione di questi casi ha condotto alla definizione di “GH bioinattivo” che è responsiva al trattamento con GH

59
Alterazioni dell’asse GH-IGF-1
GHRH GH IGF-1 Ipotalamo Ipofisi Tessuti periferici Alterazioni dell’asse GH-IGF-1 Recettore GHRH Recettore GH Recettore IGF-1

60
Regione extracellulare Regione citoplasmatica
Recettore del GH Regione extracellulare Membrana plasmatica Regione citoplasmatica

61
Recettore del GH Segnale extracellulare citoplasma GH-BP GH GHR GH GH
JAK2 JAK2 JAK2 JAK2 JAK2 JAK2 citoplasma Segnale

62
Sindrome da Insensibilità al GH
Laron nel 1966 descrisse per la prima volta tre fratelli con segni clinici e laboratoristici di deficit di GH in presenza concentrazioni estremamente elevate di GH immunoreattivo Successive descrizioni hanno contribuito a chiarire l’eziopatogenesi di questa sindrome caratterizzata da: fenotipo particolare GH basale e dopo stimolo più elevato del normale IGF-1 e IGFBP-3 bassi o indosabili nessuna risposta al test di generazione dell’ IGF-1 Il difetto molecolare risiede nel gene del recettore del GH (GHR).
.")
63
Ipoplasia mediana del viso nella sindrome di Laron:
effetto del severo deficit di IGF-1 Laron et al. Isr J Med Sci, 1966

64
Difetti del recettore del GH
Tipo selvaggio Laron S. Difetti citoplasmatici GH-BP JAK2 GH extracellulare citoplasma Segnale No Segnale No Segnale No Segnale

65
Criteri diagnostici della Sindrome da Insensibilità al GH (GHIS)
Parametri Criteri Punteggio Altezza (SDS) < GH basale (ng/ml) > Risposta allo stimolo normale o esagerata IGF-1 (ng/ml) < IGF-BP < 2 SD GH-BP (%) < IGF-1 generation test IGF-1 (ng/ml) incremento < IGFBP3 (mg/L) incremento < > 5
 < GH basale (ng/ml) > Risposta allo stimolo normale o esagerata. IGF-1 (ng/ml) < IGF-BP3 < 2 SD. GH-BP (%) < IGF-1 generation test. IGF-1 (ng/ml) incremento < IGFBP3 (mg/L) incremento < > 5.")
66
Alterazioni dell’asse GH-IGF-1
Ipotalamo Alterazioni dell’asse GH-IGF-1 GHRH Ipofisi Recettore GHRH GH Tessuti periferici Recettore GH IGF-1 Recettore IGF-1 Tessuti periferici

67
Struttura dell’IGF-1 C1 B30 C12 A1 D1 S S A21 B2 S S S S ARG SER SER
GLY TYR B30 ALA GLY TYR PRO PRO GLN LYS C12 ASN THR ALA A1 LYS PRO GLY PHE SER LYS ILE TYR ALA ARG LEU VAL PHE LEU ARG ASP LEU PRO D1 ASP GLY CYS GLU S S ALA MET A21 ARG GLU SER TYR CYS B2 CYS ARG ASP S S CYS PHE ARG GLY PRO CYS S S VAL GLU PHE THR LEU GLN LEU CYS ALA GLY ALA GLU LEU VAL ASP

68
Difetti del gene dell’IGF-1
Prima descrizione di Woods in un paziente di 15 anni con IUGR, difetti neurosensoriali e ritardo mentale Ridotta crescita sin dai primi mesi di vita, in assenza di risposta al trattamento con GH Test da stimolo di GH (ng/ml): basale picco insulina 2, notturno alti picchi, non dosabili IGF-1 sierico non dosabile (basale, IGF-1 generation test) IGFBP-3 normale Woods KA NEJM, 1996
: basale picco. insulina 2,5 61. notturno alti picchi, non dosabili. IGF-1 sierico non dosabile (basale, IGF-1 generation test) IGFBP-3 normale. Woods KA NEJM,")
69
Difetti del gene dell’IGF-1
L’analisi del gene IGF-1 rivela una delezione parziale che coinvolge gli esoni 4 e 5 con produzione di un peptide IGF-1 troncato Questo caso suggerisce: - il ruolo predominante dell’IGF-1 nell’accrescimento fetale - il possibile coinvolgimento nello sviluppo del SNC SER LYS ALA PRO LEU GLU ARG CYS ASP PHE TYR MET S Woods KA NEJM, 1996

70
GHRH GH IGF-1 Ipotalamo Ipofisi Tessuti periferici Recettore IGF-1

71
Difetti del recettore dell’ IGF-1
Resistenza all’IGF-1 Difetti del recettore dell’ IGF-1 Paziente prepubere Alla nascita: peso 1420 g (IUGR) Bassa statura armonica, scarso accrescimento postnatale, assenza di caratteri dismorfici Disordine ossessivo-compulsivo Picco di GH: 51ng/ml (112 mU/L) IGF -1: 891,1138 ng/ml Abuzzabab MJ et al, 2000
 Bassa statura armonica, scarso accrescimento postnatale, assenza di caratteri dismorfici. Disordine ossessivo-compulsivo. Picco di GH: 51ng/ml (112 mU/L) IGF -1: 891,1138 ng/ml. Abuzzabab MJ et al,")
72
Difetti del recettore dell’IGF-1
Recettore dell’IGF-1 nel paziente descritto: presenza di mutazioni in eterozigosi che interferiscono con il legame dell’IGF-1 al suo recettore Abuzzahab et al., NEJM, 2003

73
Deficit di GH da cause acquisite
Traumi perinatali Traumi cranici Infezioni del SNC Tumori primitivi dell’ipofisi-ipotalamo (craniofaringioma, glioma/astrocitoma, germinoma, etc) Tumori secondari (istiocitosi, linfomi, etc) Radioterapia del cranio Infiammazione autoimmune dell’ipofisi Transitorio (per-puberale, deprivazione psico-sociale)
 Tumori secondari (istiocitosi, linfomi, etc) Radioterapia del cranio. Infiammazione autoimmune dell’ipofisi. Transitorio (per-puberale, deprivazione psico-sociale)")
74
TERAPIA DEL DEFICIT DI GH

75
Indicazioni per il trattamento
Deficit di GH Indicazioni per il trattamento Deficit a patogenesi ipofisaria: picco di GH < 10 ng/ml in almeno 2 test provocativi Deficit a patogenesi ipotalamica: secrezione spontanea media nelle 24 ore o, quantomeno nelle 12 ore notturne, inferiore a 3 ng/ml Deficit dell’attività biologica: bassi livelli di IGF-1 normoresponsivi al test di generazione somatomedinica in pazienti con normale secrezione somatotropa spontanea e stimolata

76
Indicazioni autorizzate per il trattamento con GH biosintetico
Deficit di GH Deficit parziale di GH Sindrome di Turner Ritardo di crescita intra-uterino Insufficienza renale cronica Sindrome di Prader-Willy

77
Utilizzo non ancora autorizzato (in Italia) per la prescrizione della terapia con GH
Varianti normali della crescita / bassa statura idiopatica Displasie scheletriche Sindrome di Noonan Altre sindromi

78
Deficit di GH dose raccomandata hGH 0,025 mg/kg/die sottocute

79
Durata del trattamento
Il trattamento con GH biosintetico deve protrarsi fino al raggiungimento della statura definitiva al termine dell’epoca puberale È necessario sottoporre il paziente ad attento follow-up sia per la valutazione del beneficio che dei rischi connessi alla terapia La durata comunque dipende dalla eziologia e la terapia andrebbe continuata fino all’età adulta nei casi in cui sia confermata l’esistenza di un grave deficit secondo I criteri applicabili negli adulti

80
Sicurezza del trattamento
Dose farmacologica Anticorpi Intolleranza glucidica/iperinsulinismo Iperlipidemia Ipertensione Sindrome con fragilità cromosomica Leucemia/neoplasie solide

81
Displasie ossee: Acondroplasia
bassa statura disarmonica (rizomelia) mutazione del recettore 3 del fattore di crescita firbobalstico (rGFG 3): in più del 98% dei casi Glu380Arg autosomica dominante Craniomegalia, protusione delle bozze frontali, naso a sella, ipoplasia del mascellare con prognatismo mandibolare, palato ogivale Cifoscoliosi, iperlordosi lombare, lassità ligamentosa Mani e piedi: acromicria, alterazioni del numero delle falangi o clinodattilia Varismo o valgismo femoro-tibiale, lassità ligamentosa Ritardo mentale (in alcuni casi) Altezza durante l’infanzia -5 e -6 SDS Altezza da adulto: maschio 131 cm, femmina 123 cm
 mutazione del recettore 3 del fattore di crescita firbobalstico (rGFG 3): in più del 98% dei casi Glu380Arg. autosomica dominante. Craniomegalia, protusione delle bozze frontali, naso a sella, ipoplasia del mascellare con prognatismo mandibolare, palato ogivale. Cifoscoliosi, iperlordosi lombare, lassità ligamentosa. Mani e piedi: acromicria, alterazioni del numero delle falangi o clinodattilia. Varismo o valgismo femoro-tibiale, lassità ligamentosa. Ritardo mentale (in alcuni casi) Altezza durante l’infanzia -5 e -6 SDS. Altezza da adulto: maschio 131 cm, femmina 123 cm.")
82
Displasie ossee: acondroplasia

83
Displasie ossee Ipocondroplasia
bassa statura disarmonica autosomica dominante, mutazione eterozigotica di rGFG 3 (Asn540Lys nel 40-50%) Normalità del viso, craniomegalia relativa Brevità dei segmenti prossimali degli arti Brachidattilia, accentuazione lordosi lombare QI a volte ridotto Altezza da adulto: maschio 131 cm, femmina 123 cm
 Normalità del viso, craniomegalia relativa. Brevità dei segmenti prossimali degli arti. Brachidattilia, accentuazione lordosi lombare. QI a volte ridotto. Altezza da adulto: maschio 131 cm, femmina 123 cm.")
84
Displasie ossee: ipocondroplasia

85
Displasie ossee: ipocondroplasia
ipocondroplasia moderatamente severa in un giovane adulto (altezza finale 125 cm)
")
86
Displasie ossee: ipocondroplasia
ipocondroplasia lieve in un bambino con bassa statura disarmonica

87
Sindrome di Turner La sindrome di Turner è causata dall’assenza parziale o completa di uno dei cromosomi X Incidenza pari a circa 1 su 2500 neonate Principali caratteristiche: bassa statura insufficienza ovarica malformazioni cardiache e renali aspetto dismorfico La lunghezza alla nascita è normale ma la velocità di crescita tende a diminuire divenendo patologica tra i 4 e i 5 anni L’altezza finale delle ragazze Turner europee risulta attorno ai cm

88
Sindrome di Turner: caratteristiche
Incidenza Bassa statura Disgnesia gonadica Linfedema Cariotipo: 45,X (50%) 46,Xi (Xq) 45,X/46, XX
 46,Xi (Xq) 45,X/46, XX.")
89
Terapia e follow-up La terapia sostitutiva con steroidi è indicata per indurre la comparsa di caratteri sessuali secondari È stata inoltre dimostrata l’efficacia della terapia sostitutiva con ormone della crecsita (GH) che determinerebbe un incremento di circa 5-6 della statura finale La terapia con GH è in genere eseguita utilizzando un dosaggio doppio a quello utilizzato nel deficit (circa 1 U/kg/settimana)
 che determinerebbe un incremento di circa 5-6 della statura finale. La terapia con GH è in genere eseguita utilizzando un dosaggio doppio a quello utilizzato nel deficit (circa 1 U/kg/settimana)")
90
Sindrome di Turner: normalizzazione dell’altezza dopo 7 anni di terapia con GH
Gruppo B n=22 Gruppo C n=21 200 180 160 140 120 100 80 60 40 Gruppo A Età cronologica (aa) Altezza (cm) In this European study, Sas and colleagues evaluated 7-year growth data from 65 young girls with Turner syndrome (TS) receiving 3 different GH dosages. Patients with a chronologic age of 2–11 years, height < the 50th percentile for normal (Dutch) girls, and normal thyroid function were assigned randomly to receive GH mg/kg daily (group A, n=22); mg/kg daily for 1 year, followed by mg/kg daily (group B, n=22); or mg/kg daily for the first year, mg/kg daily for the second year, and 0.09 mg/kg daily thereafter (group C, n=21). Doses were adjusted for body surface area every 3 months. GH treatment was continued until adult height was reached <1 cm growth over 6 months or the patient elected to discontinue treatment (due to satisfaction with attained height). Estrogen therapy was not initiated until patients had received at least 4 years of GH and reached 12 years of age. These graphs show the individual heights of patients at baseline (dark [purple] points) and after 7 years of treatment (light [yellow] points) in each dosage group. Solid and dashed curves represent reference curves for healthy Dutch girls and untreated girls with TS, respectively. Following 7 years of GH, 55 of 65 patients (85%) achieved a height within the normal range for healthy girls, while 10 girls (15%) had heights just below the third percentile. Height SD scores were significantly higher in groups B and C compared to A (not shown). No adverse metabolic effects were reported in any dosage group during the treatment period. Sas TCJ, et al. J Clin Endocrinol Metab 1999;84:4607–4612. Altezza prima del GH Altezza dopo GH Gruppo A mg GH/kg/die Gruppo B 0,045 mg GH/kg/die per 1 anno; poi 0,0675 mg GH/kg/die Gruppo C 0,045 mg GH/kg/die per 1 anno; poi 0,.065 per il secondo anno; poi 0,09 mg GH/kg/die Sas TCJ, J Clin Endocrinol Metab, 1999
 Altezza (cm) In this European study, Sas and colleagues evaluated 7-year growth data from 65 young girls with Turner syndrome (TS) receiving 3 different GH dosages. Patients with a chronologic age of 2–11 years, height < the 50th percentile for normal (Dutch) girls, and normal thyroid function were assigned randomly to receive GH mg/kg daily (group A, n=22); mg/kg daily for 1 year, followed by mg/kg daily (group B, n=22); or mg/kg daily for the first year, mg/kg daily for the second year, and 0.09 mg/kg daily thereafter (group C, n=21). Doses were adjusted for body surface area every 3 months. GH treatment was continued until adult height was reached <1 cm growth over 6 months or the patient elected to discontinue treatment (due to satisfaction with attained height). Estrogen therapy was not initiated until patients had received at least 4 years of GH and reached 12 years of age. These graphs show the individual heights of patients at baseline (dark [purple] points) and after 7 years of treatment (light [yellow] points) in each dosage group. Solid and dashed curves represent reference curves for healthy Dutch girls and untreated girls with TS, respectively. Following 7 years of GH, 55 of 65 patients (85%) achieved a height within the normal range for healthy girls, while 10 girls (15%) had heights just below the third percentile. Height SD scores were significantly higher in groups B and C compared to A (not shown). No adverse metabolic effects were reported in any dosage group during the treatment period. Sas TCJ, et al. J Clin Endocrinol Metab 1999;84:4607–4612. Altezza prima del GH. Altezza dopo GH. Gruppo A mg GH/kg/die. Gruppo B 0,045 mg GH/kg/die per 1 anno; poi 0,0675 mg GH/kg/die. Gruppo C 0,045 mg GH/kg/die per 1 anno; poi 0,.065 per il secondo anno; poi 0,09 mg GH/kg/die. Sas TCJ, J Clin Endocrinol Metab,")
91
Sindrome di Down trisomia 21
più frequente causa genetica di ritardo mentale fenotipo neonatale: facies con profilo piatto, ipotonia, eccesso di cute sulla nuca, solco palmare unico evidenziazione successiva di altri caratteri dismorfici a carico di mani e piedi, della facies, degli occhie e del ritardo mentale il ritardo di crescita è in genere non evidente alla nascita e fino ai 6 mesi, mentre si evidenzia progressivamente fino a diventare pronunciato dai 5-6 anni di vita

92
Sindrome di Down

93
Sindrome di Down e ormone della crescita
basse concentrazioni endogene di GH normali valori di GH dopo stimolo farmacologico bassi livelli di IGF-1 tentativi di terapia con GH per 36 mesi: incremento della velocità di crescita senza effetti sul ritardo mentale poiché il rischio di leucemia è volte superiore alla popolazione normale non è considerata opportuna la terapia di GH per l’associazione non chiara di alti livelli circolanti di IGF-1 con alcuni tumori

94
Quando sospettare la Sindrome di Noonan
viso: impianto basso delle orecchie ipertelorismo ptosi miopia bassa statura difetti cardiaci difficoltà di alimentazione (76%) anormalità del visus (94%) criptorchidismo (77%) difetti della coagulazione (56%)
 anormalità del visus (94%) criptorchidismo (77%) difetti della coagulazione (56%)")
95
Quando sospettare la Sindrome di Williams
Incidenza: 1: nati Capelli neri, ricci Restringimento bitemporale Sopraciglie rade nel terzo distale Epicanto Iride stellata Naso corto con narici anteverse Radice nasale infossata Ipoplasia malare Filtro lungo Bocca grande, con labbra spesse Anormalità dentarie Padiglioni auricolari dismorfici Faccia da elfino

96
CAUSE DI BASSA STATURA MALATTIE ENDOCRINE DISPLASIE OSSEE
ANOMALIE CROMOSOMICHE E GENICHE RITARDO DI CRESCITA INTRAUTERINO MALATTIE CRONICHE SISTEMICHE BASSA STATURA PSICOSOCIALE VARIANTI NORMALI DI CRESCITA (ritardo costituzionale di crescita, bassa statura familiare)
")
97
Ritardo di crescita intrauterino (intrauterine growth retard, IUGR)
La definizione di neonato IUGR indica un minor potenziale di crescita fetale conseguente a processi patologici di origine materna, placentare o fetale. Per documentare il ritardo di accrescimento intrauterino si utilizzano rilevazioni ecografiche seriate che possono indicare il periodo di vita intrauterina durante la quale ha agito la noxa patogena dando origine ad un IUGR simmetrico o asimmetrico.

98
SGA: small for gestational age
Peso alla nascita < 10° (o al 5°) percentile per l’ età gestazionale Peso alla nascita < 2.5 Kg per l’ età gestazionale 37 settimane Peso alla nascita <2SD al di sotto del valore medio per l’ età gestazionale
 percentile per l’ età gestazionale. Peso alla nascita < 2.5 Kg per l’ età gestazionale 37 settimane. Peso alla nascita <2SD al di sotto del valore medio per l’ età gestazionale.")
99
CATCH UP GROWTH La maggioranza dei neonati IUGR (70-80%) presenta una crescita di recupero (catch-up growth) a 6-12 mesi di vita, o almeno entro i primi due anni. 20 - 40 - 60 - 80 - 100 - 3 6 12 24 Prematuri Nati a termine Percentuale (%) Età (mesi) Hokken-Koelega CA, Pediatr Res 1995
 presenta una crescita di recupero (catch-up growth) a 6-12 mesi di vita, o almeno entro i primi due anni Prematuri. Nati a termine. Percentuale (%) Età (mesi) Hokken-Koelega CA, Pediatr Res")
100
Crescita fetale Normale disponibilità di glucosio e aminoacidi GH
IGFBP-3 IGFBP-1 GH IGF-1 Insulina Il controllo della crescita fetale è affidato prevalentemente all’IGF-1 che, in presenza di normale disponibilità di glucosio e aminoacidi, garantisce un normale trasporto di glucosio ai muscoli e al cervello CRESCITA Normale trasporto di glucosio ai muscoli e al cervello

101
“Fetal salvage” GH IGF-1 Insulina IUGR
Ridotta disponibilità di glucosio e aminoacidi IGFBP-3 IGFBP-1 GH IGF-1 Insulina In caso di ridotta disponibilità di substrati viene preferzialmente salvaguardata l’attività cerebrale a discapito dei muscoli per cui si viene a determinare un ridotto potenziale di crescita fetale IUGR Ridotto trasporto nei muscoli e normale nel cervello

102
Livelli ormonali nei neonati
IGF-1 (mcg/L) Insulina (mU/L) IGFBP-3 (mcg/L) IGFBP-1 (mcg/L) GH (mcg/L) La diapositiva mostra i ridotti livelli di IGF-1, insulina e IGFBP-3 e gli aumentati livelli di IGFBP-1 e GH nei neonati IUGR rispetto ai controlli Controlli IUGR de Zegher F, Acta Paediatr 1997
 Insulina (mU/L) IGFBP-3 (mcg/L) IGFBP-1 (mcg/L) GH (mcg/L) La diapositiva mostra i ridotti livelli di IGF-1, insulina e IGFBP-3 e gli aumentati livelli di IGFBP-1 e GH nei neonati IUGR rispetto ai controlli. Controlli. IUGR. de Zegher F, Acta Paediatr")
103
Questioni aperte Variabilità della risposta al trattamento
alcune segnalazioni suggeriscono che l’IGF-1 potrebbe avere un valore predittivo Frank GR, Clin Endocrinol 1996 Effetti collaterali il GH potrebbe aggravare l’insulino-resistenza Rischio di sviluppare un diabete mellito di tipo 2 Hofman PL, J Clin Endocrinol Metab 1997 Chernausek SD, Horm Res 2004 Tuttavia… Alcuni studi suggeriscono la immediata reversibilità all’interruzione della terapia L’aumento di massa muscolare potrebbe proteggere dalla sindrome metabolica Hokken-Koelega AC, Horm Res 2003 Sas T, Clin Endocrinol 2001

104
CAUSE DI BASSA STATURA MALATTIE ENDOCRINE DISPLASIE OSSEE
ANOMALIE CROMOSOMICHE E GENICHE RITARDO DI CRESCITA INTRAUTERINO MALATTIE CRONICHE SISTEMICHE BASSA STATURA PSICOSOCIALE VARIANTI NORMALI DI CRESCITA (ritardo costituzionale di crescita, bassa statura familiare)
")
105
MALATTIE CRONICHE SISTEMICHE
malattia cronica farmaci flogosi malnutrizione Alterazioni ormonali a carico dell’asse GH-IGF-1 La malattia cronica attraverso la flogosi, la malnutrizione o l’utilizzo di farmaci che interferiscono con l’asse GH-IGF-1, possono causare uno scarso accrescimento. In genere, rispetto alle altre cause di crescita patologica, in questi casi la curva di crescita mostra una netta deflessione o un vero e proprio arresto in corrispondenza dell’esordio della patologia Scarso accrescimento

106
Malattia celiaca

107
Infiammazione cronica, malnutrizione, arresto della crescita
e pubertà ritardata nel morbo di Crohn

108
CAUSE DI BASSA STATURA MALATTIE ENDOCRINE DISPLASIE OSSEE
ANOMALIE CROMOSOMICHE E GENICHE RITARDO DI CRESCITA INTRAUTERINO MALATTIE CRONICHE SISTEMICHE BASSA STATURA PSICOSOCIALE VARIANTI NORMALI DI CRESCITA (ritardo costituzionale di crescita, bassa statura familiare)
")
109
Bassa statura da deprivazione affettiva o bassa statura psico-sociale
Vi è una stretta relazione fra crescita e stato socio-economico del bambino. La bassa statura psico-sociale è caratterizzata da: comparsa in età superiore ai due anni disturbi del comportamento polifagia polidispia non sempre è associata a deficit di GH il deficit di GH, quando associato, reversibile con il miglioramento della condizione psicosociale

110
CAUSE DI BASSA STATURA MALATTIE ENDOCRINE DISPLASIE OSSEE
ANOMALIE CROMOSOMICHE E GENICHE RITARDO DI CRESCITA INTRAUTERINO MALATTIE CRONICHE SISTEMICHE BASSA STATURA PSICOSOCIALE VARIANTI NORMALI DI CRESCITA (ritardo costituzionale di crescita, bassa statura familiare)
")
111
Ritardo costituzionale di crescita
190 180 170 160 150 140 130 età cronologica (anni) Altezza (cm) Età cronologica Età ossea
 Altezza. (cm) Età cronologica. Età ossea.")
112
QUANDO SOSPETTARE LA BASSA STATURA FAMILIARE
aspetto armonico obiettività nella norma familiarità per bassa statura target zone ± 8.5 cm curva di crescita parallela ma inferiore al 3° centile Velocità di crescita normale età ossea = età cronologica

Presentazioni simili

















>")

 PER CONTROLLARE L’ATTIVITA’ DI ALTRI ORGANI AD UNA CERTA DISTANZA L’ORMONE: PRODOTTO.>")