Scaricare la presentazione
1
SINDROMI

2
SINDROME DI ANGELMAN Nel 1965 Harry Angelman, pediatra inglese, descrisse una sindrome che chiamò “happy puppet syndrome” caratterizzata da ritardo mentale anche grave, scoppi incomprensibili di riso, particolare aspetto del volto, atassia, movimenti a scatti di gambe e braccia.

4
I bambini che ne sono affetti hanno in genere un temperamento allegro e un carattere socievole, con un’apparenza opposta a quella contraddistinta dal difetto di interazione intenzionale e abilità comunicative tipica dell’autismo

5
Si presume che l’incidenza (frequenza rispetto alle nascite attuali) sia fra 1:12.000 e 1:30.000. Non vi sono differenze significative fra maschi e femmine.

6
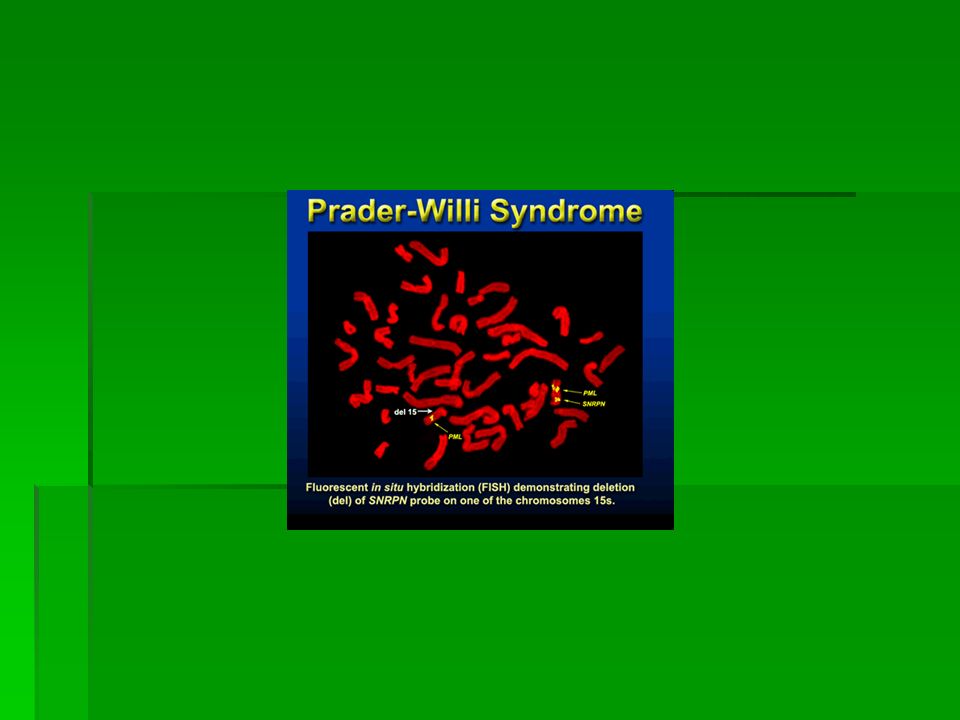
genetica La sindrome è caratterizzata da delezione sul braccio lungo del cromosoma 15 (15q11-q13). Anche la sindrome di Prader-Willi è dovuta ad una delezione nel cromosoma 15. Mentre in questo secondo caso il cromosoma coinvolto è quello di origine paterna, nella sindrome di Angelman è quello di origine materna.

7
Spieghiamoci meglio questa sindrome viene spesso citata come esempio di imprinting genetico in quanto generalmente causato da delezione o inattivazione di geni del cromosoma 15 ereditato dalla madre. La sindrome sorella, causata da imprinting materno, è la Prader- Willi. Molti degli elementi caratteristici della sindrome di Angelman derivano dalla perdita di funzione del gene Ube3A. Normalmente si eredita una copia del gene da ciascun genitore ed entrambe le copie sono attive in molti tessuti del corpo, ma in alcune aree del cervello è attiva solo la copia ereditata dalla madre. Questa attivazione specifica del gene di un genitore è causata da un fenomeno noto come genomic imprinting. Se la copia materna del gene Ube3A è perduta a causa di un’alterazione cromosomica o di una mutazione genica, una persona non avrà copie attive in alcune parti del cervello. Più in generale, l’imprinting genomico è definito come un processo che conferisce un’impronta specifica ai geni provenienti da ciascun genitore, così che i geni imprinted provenienti dalla madre saranno diversi e riconoscibili da quelli trasmessi dal padre. In pratica si parla di imprinting genetico o genomico quando l’espressione di un gene o di un gruppo di geni dipende da un tratto del genoma che proviene solo dal padre o solo dalla madre.

9
clinica Due terzi dei soggetti con sindrome di Angelman hanno occhi azzurri e capelli biondi. La sindrome comporta, di norma, movimenti di tipo atassico, a scatti, volto allungato, mandibola prominente, bocca ampia con denti spaziati, labbro superiore sottile, occhi infossati, occipite piatto. Spesso è presente microcefalia. In circa due terzi dei casi vi sono difficoltà di suzione e deglutizione e rigurgiti frequenti. Lo sviluppo motorio è ritardato. Singolare è la frequenza di scoppi di riso immotivati Frequenti i disturbi del sonno Presenti anomalie nel tracciato elettroencefalografico con epilessia in più dell’80% dei casi

10
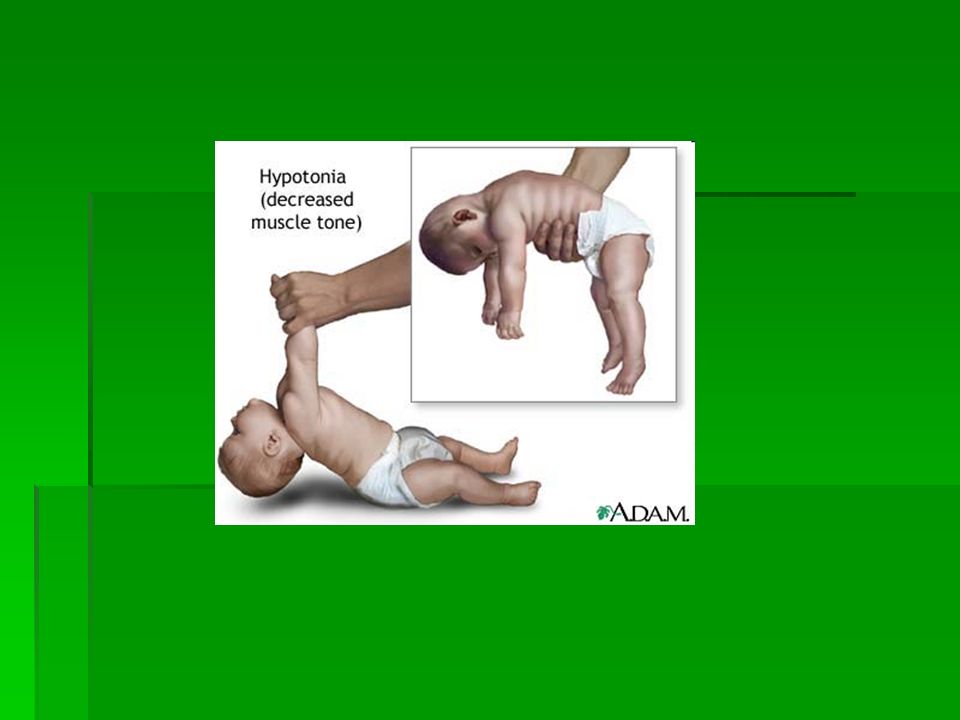
SINDROME DI PRADER- WILLI colpisce un bambino ogni 15.000 nati I neonati, a causa di uno scarso tono muscolare (ipotonia), generalmente si nutrono e succhiano con difficoltà e spesso devono essere alimentati con biberon speciali o sonde. Riescono a stare seduti, strisciare e camminare più tardi dei loro coetanei. Tra i 2 o 4 anni molti di loro sviluppano un appetito insaziabile che può degenerare in un'obesità eccessiva se non viene subito controllata. Questo appetito, accompagnato da un facile aumento di peso, problemi di comportamento e temperamento testardo ed irascibile sono i maggiori problemi associabili alla sindrome.

12
Nei primi anni di vita i bambini con sindrome di Prader-Willi hanno un carattere gioviale ed allegro e solitamente non presentano problemi di comportamento. La maggior parte di ragazzi ed adulti con PWs ha invece difficoltà comportamentali, che coincidono con l'insorgenza di iperfagia (anche se non tutti i problemi di comportamento sono da correlare al cibo) e che presentano un picco di sintomatologia nell'adolescenza e nell'età adulta giovanile.
 e che presentano un picco di sintomatologia nell adolescenza e nell età adulta giovanile..")
13
Lo sviluppo puberale, in entrambi i sessi, è quasi sempre del tutto assente o ritardato ed è presente un ritardo mentale di grado variabile.

14
La malattia è invalidante ed ha un percorso cronico senza speranza di guarigione. I pazienti devono essere assiduamente sorvegliati e sono necessari frequenti controlli medici. E’ possibile intervenire favorevolmente con adeguate misure di prevenzione, di educazione alimentare e di riabilitazione.

16
SINDROME DI WILLIAMS è una rara malattia genetica conosciuta dal 1961,quando il Dr. J.C.P. Williams del Green Lane Hospital di Ankland (Nuova Zelanda) e collaboratori pubblicarono i dati clinici di quattro pazienti, caratterizzati da aspetti del volto simili,stenosi aortica sopravalvolare,ritardo psicomotorio e scarso accrescimento staturo-ponderale. Un anno più tardi (1962) Breuen e collaboratori pubblicarono i dati clinici di altri quattro pazienti con le stesse caratteristiche, inoltre i pazienti descritti presentavano un carattere particolarmente socievole ed estroverso.
 e collaboratori pubblicarono i dati clinici di quattro pazienti, caratterizzati da aspetti del volto simili,stenosi aortica sopravalvolare,ritardo psicomotorio e scarso accrescimento staturo-ponderale. Un anno più tardi (1962) Breuen e collaboratori pubblicarono i dati clinici di altri quattro pazienti con le stesse caratteristiche, inoltre i pazienti descritti presentavano un carattere particolarmente socievole ed estroverso..")
17
La SW è una patologia data da una alterazione del patrimonio genetico localizzata sul braccio lungo del cromosoma 7 (7q11.23).All'interno di questa regione è certamente localizzato un gene che regola la produzione di una importante proteina : l'Elastina. Questa proteina rappresenta la costituente principale del tessuto connettivo elastico la cui mancata produzione provoca gravi problemi a carico del cuore e di altri organi, influendo anche sull’aspetto fisico dei piccoli pazienti.

18
clinica Microcefalia con fronte ampia. Gli occhi appaiono particolarmente distanziati tra loro (ipertelorismo), con presenza di strabismo e di difetti visivi che interessano la rifrazione e il senso della profondità. La radice del naso è infossata, le labbra grosse e il mento piccolo. I denti possono risultare mal disposti, in numero inferiore al normale (oligodontia), di dimensioni più piccole (microdontia) e con carenze allo smalto e predisposizione alla carie. Le spalle, poi, hanno un aspetto cadente e il collo appare allungato. Molto comuni sono anche i problemi cardiologici (in particolare la stenosi sopravalvolare aortica e quella polmonare) e disturbi quali iperacusia, ipercalcemia, complicazioni renali e vescicali, ipotono muscolare, problemi gastrointestinali e pubertà precoce.
, con presenza di strabismo e di difetti visivi che interessano la rifrazione e il senso della profondità. La radice del naso è infossata, le labbra grosse e il mento piccolo. I denti possono risultare mal disposti, in numero inferiore al normale (oligodontia), di dimensioni più piccole (microdontia) e con carenze allo smalto e predisposizione alla carie. Le spalle, poi, hanno un aspetto cadente e il collo appare allungato. Molto comuni sono anche i problemi cardiologici (in particolare la stenosi sopravalvolare aortica e quella polmonare) e disturbi quali iperacusia, ipercalcemia, complicazioni renali e vescicali, ipotono muscolare, problemi gastrointestinali e pubertà precoce..")
19
... ritardo mentale, e nello sviluppo motorio, di livello variabile dal medio al grave. Le conseguenti difficoltà di apprendimento e di coordinazione motoria non impediscono lo sviluppo di una certa espansività, socievolezza e capacità di espressione.

21
RICORDARE CHE NELLE VARIE SINDROMI non tutte le caratteristiche sono presenti in ogni soggetto affetto e la loro gravità può variare in modo significativo









>")














