Scaricare la presentazione
1
Risonanza magnetica nucleare
Si ricavano informazioni dallo studio delle proprietà magnetiche dei nuclei

2
Lo spin nucleare Lo spin è una proprietà fondamentale come la carica e la massa. Protoni, elettroni e neutroni possiedono uno spin. Molti nuclei atomici si comportano come delle particelle cariche che ruotano intorno al proprio asse. Il momento angolare o momento di spin P che ne risulta è ovviamente quantizzato. P è un vettore orientato lungo l’asse della rotazione, e il numero di spin I è un indice dell’intensità (o modulo) di questo momento angolare. P = I= numero di spin valori=0, ½, 1, e così via fino a 6.
 di questo momento angolare. P = I= numero di spin valori=0, ½, 1, e così via fino a 6.")
3
Il modulo del momento di spin P è fisso per ogni nucleo, e non può cambiare in nessun modo.
Può cambiare l’ orientazione che il vettore momento di spin può assumere nei confronti di una direzione esterna z (per esempio, nell’NMR, quella del campo magnetico applicato). Anche questa orientazione è quantizzata, ed il vettore momento di spin può assumere solo 2I+1 orientazioni definite dal numero quantico di spin m. Pz = m Il numero quantico m può assumere i valori ‑I,-I+1,….., +I-1, +I.
. Anche questa orientazione è quantizzata, ed il vettore momento di spin può assumere solo 2I+1 orientazioni definite dal numero quantico di spin m. Pz = m. Il numero quantico m può assumere i valori ‑I,-I+1,….., +I-1, +I.")
4
Numeri di spin di alcuni nuclei
Elemento H 2H 12C 13C 14N 16O 17O 19F Numero quantico di spin ( I ) 1/ / /2 1/2 Orientazioni Elementi con numero di massa o numero atomico dispari sono dotati di “spin nucleare”. Gli isotopi più abbondanti del C e dell’ O non possiedono spin.
 1/ / /2 1/2. Orientazioni Elementi con numero di massa o numero atomico dispari sono dotati di spin nucleare . Gli isotopi più abbondanti del C e dell’ O non possiedono spin.")
5
Momento magnetico nucleare
Poiché il nucleo atomico è carico, e ogni carica in movimento genera un campo magnetico, ogni nucleo dotato di spin si comporterà come un piccolo magnete, cioè sarà dotato di un momento magnetico m. Il momento magnetico m è proporzionale al momento di spin e ne ha la stessa direzione: quindi il suo modulo non può variare, e può assumere solo due possibili orientazioni per un nucleo con I = ½. La costante di proporzionalità tra il momento magnetico m ed il momento angolare nucleare P è detta costante giromagnetica m m = g P + m g= rapporto giromagnetico mz = g Pz = m g

6
B0 Energia di un nucleo N -1/2 +1/2 S
In assenza di un campo magnetico esterno, i vari possibili stati di spin hanno la stessa energia, e non è quindi possibile alcuna interazione con la radiazione elettromagnetica. Tuttavia, se il nucleo è messo in presenza di un campo magnetico, i due stati di spin hanno diversa energia: se il momento magnetico m è “parallelo” (nucleo nello stato +½ o a) al campo magnetico si ha una interazione favorevole tra i due, e l’energia del nucleo scende; se invece m è “antiparallelo” (nucleo nello stato –½ o b) l’interazione è sfavorevole e l’energia del nucleo sale. B0 +1/2 -1/2 N S
 al campo magnetico si ha una interazione favorevole tra i due, e l’energia del nucleo scende; se invece m è antiparallelo (nucleo nello stato –½ o b) l’interazione è sfavorevole e l’energia del nucleo sale. B0. +1/2. -1/2. N. S.")
7
Il Fenomeno della risonanza

8
Assorbimento di Energia
+1/2 -1/2 DE = hn DE quantizzata Radiofrequenza Allineata Opposta

9
n = DE/h L’energia di un nucleo in un campo magnetico
Poiché i due stati di spin hanno energia diversa, è quindi possibile indurre il passaggio da uno stato all’altro mediante un quanto di radiazione elettromagnetica di frequenza n = DE/h Nell’NMR la differenza di energia tra gli stati nucleari è indotta dal campo magnetico esterno, e può quindi essere variata. L’energia di un nucleo in un campo magnetico Per un nucleo con I = ½, m puo essere +½ o –½, per cui i due suoi possibili livelli energetici sono: La differenza di energia tra i due livelli nucleari è: la frequenza di assorbimento è:

10
La differenza di Energia dipende da B0 e g
- 1/2 Degeneri a B0 = 0 + 1/2 B0

11
g è una costante per ciascun nucleo (H, C, N, etc) n = 2p Bo
La frequenza di Larmor!!! g è una costante per ciascun nucleo (H, C, N, etc) poiché DE = hn Rapporto giromagnetico g Campo magnetico applicato Frequenza della radiazione che in grado di operare una transizione g n = 2p Bo
 poiché DE = hn. Rapporto giromagnetico g. Campo magnetico applicato. Frequenza della. radiazione che in. grado di operare una. transizione. g. n = 2p. Bo.")
12
La sensibilità DE<<<KT
Principale problema della spettroscopia NMR Distribuzione di Boltzmann DE<<<KT La sensibilità di un esperimento NMR aumenta all’aumentare del campo applicato; inoltre, nuclei con rapporto giromagnetico elevato sono più sensibili di nuclei con rapporto giromagnetico più basso

13
Il chemical shift In assenza di campo magnetico, gli elettroni si muovono in maniera disordinata formando una nube elettronica intorno al nucleo.

14
Il campo magnetico induce un movimento più ordinato degli elettroni, come se percorressero un orbita su un piano perpendicolare al campo magnetico.

15
Il movimento degli elettroni produce a sua volta un campo magnetico (che è quindi indotto dal campo magnetico esterno). Le linee di forza di questo campo magnetico sono indicate in rosso.

16
All’interno della nuvola elettronica, il campo magnetico indotto si oppone al campo magnetico applicato, ed il nucleo è schermato. Una nube di elettroni intorno ad un nucleo scherma il nucleo.

17
Invece, intorno alla nuvola elettronica il campo magnetico è nella stessa direzione del campo magnetico esterno. Un nucleo che si trovasse in questa zona sarebbe deschermato.

18
Il chemical shift B= B0 (1-s)
Sotto l’influenza del campo magnetico esterno gli elettroni tendono ad assumere un movimento rotatorio, e ruotando generano essi stessi un campo magnetico. Nonostante tutti i nuclei di una certo tipo (per esempio 1H) siano esattamente identici, essi risuonano a frequenze leggermente diverse purchè si trovino in intorni chimici differenti. Il campo magnetico generato si oppone al campo magnetico esterno nella zona centrale dell’orbitale, mentre si somma nella sua periferia. Poiché il nucleo si trova al centro dell’atomo, esso sarà sottoposto ad un campo magnetico effettivo minore di quello applicato, è cioè schermato dagli elettroni. B= B0 (1-s)
 siano esattamente identici, essi risuonano a frequenze leggermente diverse purchè si trovino in intorni chimici differenti. Il campo magnetico generato si oppone al campo magnetico esterno nella zona centrale dell’orbitale, mentre si somma nella sua periferia. Poiché il nucleo si trova al centro dell’atomo, esso sarà sottoposto ad un campo magnetico effettivo minore di quello applicato, è cioè schermato dagli elettroni. B= B0 (1-s)")
19
Misura del chemical shift
Dipende dal campo Indipendente dalle condizioni sperimentali Perché il TMS? dodici protoni equivalenti e molto schermati solubile nella maggior parte dei solventi organici facilmente allontanabile

20
Il chemical shift protonico
d- d+ C H Cl Elemento elettronegativo Il principale fattore che dermina il chemical shift di un protone è la densità elettronica del relativo idrogeno. Composto CH3X CH3F CH3OH CH3Cl CH3Br CH3I CH4 (CH3)4Si Elemento X F O Cl Br I H Si Elettronegatività of X Chemical shift d TMS Più deschermati Il potere descharmante aumenta con l’elettronegatività di X
4Si. Elemento X. F O Cl Br I H Si. Elettronegatività of X Chemical shift d TMS. Più deschermati. Il potere descharmante aumenta con l’elettronegatività di X.")
21
Anisotropia del chemical shift
I protoni olefinici (legati a carboni sp2) hanno chemical shifts molto più alti dei protoni legati a carboni sp3, con valori tipici di d 5-6. Ciò è dovuto alla nuvola elettronica p che può anche deschermare il protone, se la molecola è orientata opportunamente rispetto al campo magnetico. C=C H I protoni sono deschermati Linee di forza del campo magnetico indotto BO Se il piano dell’alchene è perpendicolare alla direzione del campo magnetico, gli elettroni possono muoversi efficacemente ed i protoni olefinici risultano deschermati, come si vede nella figura a destra. Perciò i protoni olefinici sono più deschermati di quanto dovrebbero essere sulla base dei soli effetti di elettronegatività.
 hanno chemical shifts molto più alti dei protoni legati a carboni sp3, con valori tipici di d 5-6. Ciò è dovuto alla nuvola elettronica p che può anche deschermare il protone, se la molecola è orientata opportunamente rispetto al campo magnetico. C=C. H. I protoni sono deschermati. Linee di forza del campo magnetico indotto. BO. Se il piano dell’alchene è perpendicolare alla direzione del campo magnetico, gli elettroni possono muoversi efficacemente ed i protoni olefinici risultano deschermati, come si vede nella figura a destra. Perciò i protoni olefinici sono più deschermati di quanto dovrebbero essere sulla base dei soli effetti di elettronegatività.")
22
H C Alchini Bo I protoni sono schermati
Densità elettronica a simmetria cilindrica intorno all’asse del legame CC. Come si può vedere i protoni si trovano in una zona in cui il campo magnetico generato dagli elettroni in movimento si oppone al campo magnetico esterno, per cui risultano schermati. Bo H C I protoni sono schermati Linee di forza del campo magnetico indotto

23
Corrente di anello nel benzene
elettroni p Deschermato H H B0

24
d (ppm) -OH -NH Campi alti Idrogeni schermati Campi bassi
Idrogeni deschermati CHCl3 , TMS d (ppm) 12 11 10 9 8 7 6 5 4 3 2 1 H CH2F CH2Cl CH2Br CH2I CH2O CH2NO2 CH2Ar CH2NR2 CH2S C C-H C=C-CH2 CH2-C- C-CH-C RCOOH RCHO C=C C C-CH2-C C-CH3 O
 H. CH2F. CH2Cl. CH2Br. CH2I. CH2O. CH2NO2. CH2Ar. CH2NR2. CH2S. C C-H. C=C-CH2. CH2-C- C-CH-C. RCOOH. RCHO. C=C. C. C-CH2-C. C-CH3. O.")
25
CHEMICAL SHIFTS (ppm) R-CH3 0.7 - 1.3 R-C=C-C-H 1.6 - 2.6
R-C-C-H O RO-C-C-H HO-C-C-H N C-C-H R-C C-C-H C-H R-N-C-H R-S-C-H I-C-H Br-C-H Cl-C-H RO-C-H HO-C-H R-C-O-C-H R-C=C-H H R-C-H R-C-O-H O2N-C-H F-C-H R3CH R-CH2-R R-N-H Ar-N-H R-S-H R-O-H Ar-O-H R-C-N-H R-C C-H

26
NON DOBBIAMO RICORDARE TUTTI I VALORI A MEMORIA !!!!
alifatico C-H CH vicino a un legame p C-H dove C è legato ad un atomo elettronegativo alcheni =C-H benzene CH aldeidi CHO acidi COOH 2 3 4 6 7 9 10 X-C-H X=C-C-H Molti spettri possono essere interpretati riferendoci a questo schema

27
Integrazione L’intensità di un segnale nello spettro NMR è proporzionale al numero di protoni che lo genera.

28
Acetato di benzile 55 : 22 : 33 = 5 : 2 : 3 Linea integrale
L’altezza delle linee integrali è proporzionale al numero di H di ciascun picco 55 : 22 : = : 2 : 3 Linea integrale

29
Accoppiamento di spin I segnali in uno spettro NMR hanno spesso struttura fine. La causa di questa struttura fine che rende più complessi, ma anche più ricchi di informazioni, gli spettri NMR, è l’accoppiamento spin-spin, ossia l’influenza degli stati di spin di un nucleo sulla frequenza di risonanza dei nuclei che lo circondano.

30
1,1,2-tricloroetano integrale = 2 integrale = 1 tripletto doppietto

31
Origine dell'accoppiamento
Consideriamo un sistema formato da due protoni che chiamiamo HA e HX. Il protone HA puo trovarsi negli stati di spin a e b con probabilità quasi uguale (come sappiamo le due popolazioni sono quasi identiche): Poiché ogni nucleo possiede un momento magnetico (ossia è un piccolo magnete), il campo magnetico del nucleo HX può influenzare quello subito dal nucleo HA, e come si vede dalla figura questa influenza è opposta se il nucleo HA è nello stato a o b: nel primo caso HX è schermato, nel secondo è deschermato. Di conseguenza, HX risuona a frequenze leggermente diverse se si trova vicino ad un HA nello stato a o un HA nello stato b.
: Poiché ogni nucleo possiede un momento magnetico (ossia è un piccolo magnete), il campo magnetico del nucleo HX può influenzare quello subito dal nucleo HA, e come si vede dalla figura questa influenza è opposta se il nucleo HA è nello stato a o b: nel primo caso HX è schermato, nel secondo è deschermato. Di conseguenza, HX risuona a frequenze leggermente diverse se si trova vicino ad un HA nello stato a o un HA nello stato b.")
32
La frequenza di risonanza di HA dipende dagli stati di spin dei nuclei vicini
Allineato con B0 Opposto a Bo +1/2 -1/2 50 % di molecole 50 % di molecole H H H H A A C C C C Bo doppietto

33
La costante di accoppiamento
La distanza (= differenza di frequenza) tra i due segnali del doppietto (di solito detti rami del doppietto) è detta costante di accopiamento ed indicata col simbolo J. Il chemical shift del protone che dà origine al doppietto è misurato al centro del doppietto.
 tra i due segnali del doppietto (di solito detti rami del doppietto) è detta costante di accopiamento ed indicata col simbolo J. Il chemical shift del protone che dà origine al doppietto è misurato al centro del doppietto.")
34
L’ accoppiamento spin-spin è dovuto alle interazioni tra i momenti magnetici nucleari, che sono indipendenti dal campo magnetico applicato B0 per cui le costanti di accoppiamento J sono indipendenti dal campo magnetico applicato e risultano uguali in qualunque spettrometro. Si misurano in Hz e non in ppm. 60 MHz L’ aspetto dello spettro varia al variare del campo dello spettrometro 200 MHz 600 MHz

35
Sistemi di spin di primo ordine
Consideriamo due nuclei A e X accoppiati tra loro. Quando i due nuclei hanno un chemical shift molto differente, lo spettro appare formato da due doppietti ben distinti Dn>>J AX Se i chemical shift diventano più vicini, i due doppietti iniziano a deformarsi, con i rami esterni che diventano meno intensi dei rami interni (questo è detto effetto tetto):
:")
36
Sistema A2X CHCl2–CH2Cl

37
Sistema A3X2 C H

38
La regola n+1 singoletti doppietti tripletti quartetti quintetti
Per prevedere la molteplicità singoletti doppietti tripletti quartetti quintetti sestetti eptetti Due nuclei vicini n+1 = 3 tripletto Un nucleo vicino n+1 = 2 doppietto

39
( x = y ) ( x = y )
 ( x = y )")
40
Il triangolo di Tartaglia
Intensità delle linee del multipletto 1 singletto 1 1 doppietto tripletto quartetto quintetto sestetto eptetto ottetto

41
Spettro NMR del Bromoetano

42
Spettro NMR dell'acetaldeide

43
Spettro NMR del 2-nitropropano

44
Eccezioni alla regola N+1
Protoni equivalenti per simmetria non accoppiano Nessun accoppiamento se x=y 1) 2) accoppiamenti tra protoni con chemical shift coincidenti non sono visibili or IMPORTANTE !
 2) accoppiamenti tra protoni con chemical shift coincidenti non sono visibili. or. IMPORTANTE !")
45
Eccezioni alla regola N+1
Questa regola può essere applicata se le tutte le costanti di accoppiamento sono uguali, il che succede spesso soprattutto se il protone è accoppiato a protoni equivalenti. AMX Doppio doppietto

46
Costanti di accoppiamento
Costante di accoppiamento geminale 2J 2J Protoni su carboni sp3: J Hz Protoni su carboni sp2: J 1-3 Hz 3J Costante di accoppiamento vicinale 3J.

47
3J Costante di accoppiamento vicinale 3J.
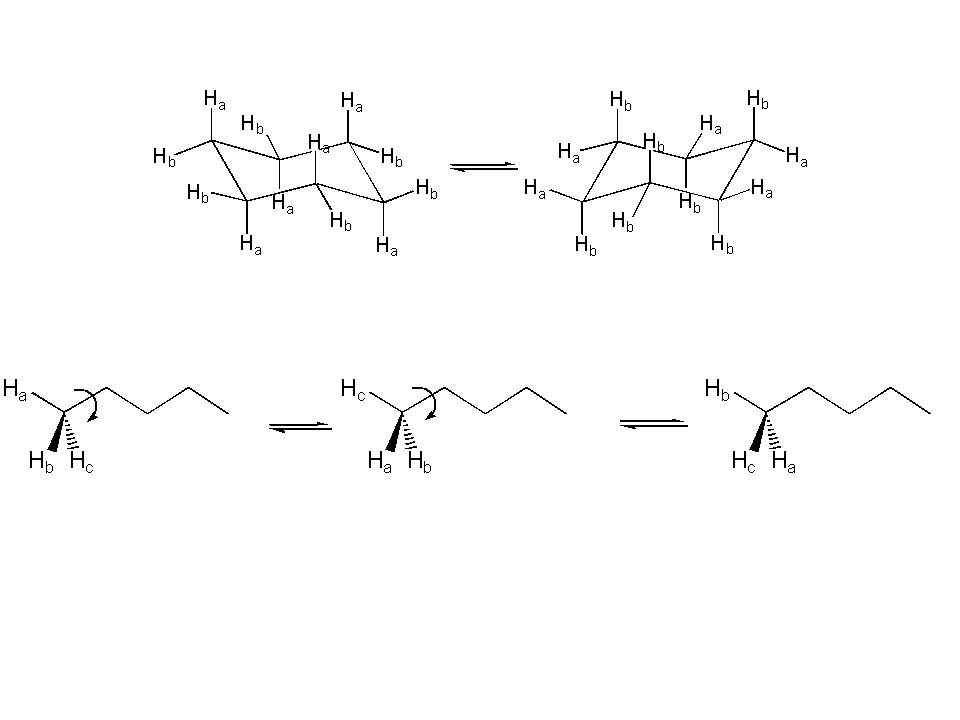
Legame singolo, libera rotazione: J 7 Hz Legame singolo, rotazione impedita: legge di Karplus Legame singolo, cicli a 6 termini (forma a sedia): Jax,ax 10 Hz Jax,eq 3 Hz Jeq,eq 3 Hz Legame singolo, cicli a 5 termini: J 5 Hz (cis,trans) Legame doppio: Jcis 7-11 Hz Jtrans Hz Composti aromatici a 6 termini: Jorto 7-9 Hz Composti aromatici a 5 termini: Jorto 3-5 Hz
: Jax,ax 10 Hz. Jax,eq 3 Hz. Jeq,eq 3 Hz. Legame singolo, cicli a 5 termini: J 5 Hz (cis,trans) Legame doppio: Jcis 7-11 Hz. Jtrans Hz. Composti aromatici a 6 termini: Jorto 7-9 Hz. Composti aromatici a 5 termini: Jorto 3-5 Hz.")
48
Costante di accoppiamento long-range.
Accoppiamento allilico (4J: CH-C=CH): J 0-2 Hz Accoppiamento omoallilico (5J: CH-C=C-CH): J 0-2 Hz Accoppiamenti meta e para in composti aromatici: Jmeta 1-3 Hz Jpara 0-1 Hz Accoppiamento W: 4J 0-7 Hz
: J 0-2 Hz. Accoppiamento omoallilico (5J: CH-C=C-CH): J 0-2 Hz. Accoppiamenti meta e para in composti aromatici: Jmeta 1-3 Hz. Jpara 0-1 Hz. Accoppiamento W: 4J 0-7 Hz.")
49
Legge di Karplus La costante di accoppiamento vicinale dipende principalmente dall’angolo diedro tra i due protoni. Come si vede nel grafico, è piccola per idrogeni sghembi (f=60°), e grande per idrogeni anti (f=180°). J = J0 cos2f per < f < 90 J = J180 cos2f per < f < 180
, e grande per idrogeni anti (f=180°). J = J0 cos2f per 0 < f < 90. J = J180 cos2f per 90 < f < 180.")
50
Accoppiamento dipolare Accoppiamento scalare
Non visibile in spettri NMR in soluzione Dipende principalmente dalla distanza tra i nuclei Costanti di accoppiamento elevate quando visibili (centinaia o migliaia di Hz) Accoppiamento scalare Visibile in spettri NMR in soluzione Dipende principalmente dal numero di legami tra i nuclei Costanti di accopiamento piuttosto piccole (decine o centinaia di Hz per le 1J, 0-20 Hz per le 2J e 3J)
 Accoppiamento scalare. Visibile in spettri NMR in soluzione. Dipende principalmente dal numero di legami tra i nuclei. Costanti di accopiamento piuttosto piccole (decine o centinaia di Hz per le 1J, 0-20 Hz per le 2J e 3J)")
51
Lo spettrometro Uno spettrometro NMR è formato da alcuni componenti fondamentali: un magnete, che deve produrre un intenso ed uniforme campo magnetico nel quale deve essere posto il campione un generatore di radiofrequenza un ricevitore di radiofrequenza.

52
RF Il campo magnetico deve essere il più uniforme
1) Il magnete superconduttore: Sono delle leghe raffreddate alla temperatura dell’elio liquido, che è solo pochi gradi al di sopra dello zero assoluto. RF Il campo magnetico deve essere il più uniforme possibile (disomogeneità < 1 parte su 109!)
 Il magnete superconduttore: Sono delle leghe raffreddate alla temperatura dell’elio liquido, che è solo pochi gradi al di sopra dello zero assoluto. RF. Il campo magnetico deve essere il più uniforme. possibile (disomogeneità < 1 parte su 109!)")
53
2) Il Trasmettitore/ricevitore di radiofrequenza:
Oppure: Consolle computer + RF (il nostro AMX-500!) 3) Il computer per il controllo ed l’elaborazione dei dati:
 3) Il computer per il controllo ed. l’elaborazione dei dati:")
54
Spettrometria NMR: aspetti tecnici
La spettroscopia NMR ad alta risoluzione va effettuata su campioni in soluzione a causa di: Effetti anisotropi sul chemical shift Accoppiamenti dipolari Solventi per l’NMR: non devono contenere protoni Molti disponibili: CDCl3, CD3OD, D2O, CD3COCD3, C6D6, …… Ma anche H2O se necessario (protoni scambiabili) con tecniche particolari Sistema di lock a deuterio: La presenza del deuterio è importante anche perché questo nucleo serve come “ancora” per mantenere costante il campo magnetico. Infatti c’è una costante irradiazione alla frequenza di risonanza del deuterio, e se il campo magnetico varia, il deuterio va fuori risonanza. Rotazione campione: La cuvetta può essere fatta ruotare sul proprio asse, per migliorare l’omogeneità del campo magnetico. In questo caso, possono comparire le cosiddette “bande laterali”. Oggi normalmente non si usa questa tecnica.
 con tecniche particolari. Sistema di lock a deuterio: La presenza del deuterio è importante anche perché questo nucleo serve come ancora per mantenere costante il campo magnetico. Infatti c’è una costante irradiazione alla frequenza di risonanza del deuterio, e se il campo magnetico varia, il deuterio va fuori risonanza. Rotazione campione: La cuvetta può essere fatta ruotare sul proprio asse, per migliorare l’omogeneità del campo magnetico. In questo caso, possono comparire le cosiddette bande laterali . Oggi normalmente non si usa questa tecnica.")
55
NMR ad onda continua Per ottenere lo spettro NMR si può:
variare con continuità la frequenza della radiazione elettromagnetica e misurare l’assorbimento per ogni frequenza (scansione della frequenza) lasciare costante la frequenza della radiazione elettromagnetica e variare con continuità il campo magnetico (scansione del campo): all’aumentare del campo magnetico entreranno in risonanza protoni via via più schermati, e si otterrà comunque uno spettro.
 lasciare costante la frequenza della radiazione elettromagnetica e variare con continuità il campo magnetico (scansione del campo): all’aumentare del campo magnetico entreranno in risonanza protoni via via più schermati, e si otterrà comunque uno spettro.")
56
NMR a impulsi (n1 ..... nn) n2 n1 n3 N S
Contiene un intervallo di frequenze RF a banda larga Con un singolo impulso sono eccitati simultaneamente tutti i protoni N S n1 n2 n3 (n nn)
")
57
FREE INDUCTION DECAY ( rilassamento )
Ogni nucleo emette una radiazione alla propria frequenza di risonanza, per cui quello che si ottiene è una sovrapposizione di onde sinusoidali a diverse frequenze.

58
FID totale spettro nel “dominio de tempo” n1 + n2 + n tempo

59
TRASFORMATA DI FOURIER
Il complesso segnale del FID è trasformato nello spettro NMR attraverso complesse operazioni matematiche DOMINIO DEL TEMPO Convertito in DOMINIO DELLE FREQUENZE FID SPETTRO NMR FT-NMR SEGNALE COMPLESSO n1 + n2 + n Frequenze individuali Un insieme di frequenze che decadono nel tempo Spettro NMR

60
Il FID è trasformato in un classico spettro NMR
spettro nel “dominio delle frequenze”

61
Aumenta la sensibilità
Vantaggi dell’ FT-NMR La fase di eccitazione, la registrazione del FID e la FT sono molto veloci richiedono solo pochi secondi Il ciclo può essere ripetuto n volte Aumenta la sensibilità

62
RAPPORTO S/R segnale rumore 1° impulso 2° impulso sommando R ≈
Aumenta S ≈ N segnale rumore 1° impulso 2° impulso sommando R ≈ nth pulse etc.

63
Protoni equivalenti Molto spesso i protoni possono risuonare alla stessa frequenza perché si trovano nello stesso intorno chimico, ossia perché sono equivalenti: per simmetria per rotazione libera

65
Alcuni semplici esercizi

66
Composto 1: un chetone C6H12O

67
Composto 2: alcol etere C8H10O2

68
Composto 3: estere C5H8O2

69
Composto 4: C4H8O

70
Composto 5: C7H7NO2

71
Composto 6: un ammide C8H9NO

72
Composto 7: un etere C4H8O

73
Composto 8: un estere C10H12O2

74
La precessione Quando un protone è posto in un campo magnetico
….. Comincia a precedere L’ asse magnetico del protone compie una precessione intorno all’asse di applicazione di B0 allo stesso modo di una trottola inclinata sotto l’effetto della gravità terrestre.

75
N w Quando sono posti in un campo magnetico, i nuclei precedono con frequenza w. RADIOFREQUENZA MHz hn NMR Se n = w il nucleo assorbirà la radiazione con conseguente inversione del suo spin S

76
Un insieme di protoni equivalenti che compiono una precessione in fase casuale lungo l’asse z (direzione di B0) producono una magnetizzazione macroscopica netta M0, detta magnetizzazione longitudinale, lungo l’asse z. Modello vettoriale

77
Quando la radiofrequenza applicata è uguale alla frequenza di precessione, i nuclei entrano in risonanza. La radiofrequenza viene generata con un oscillatore posto lungo l’asse x in modo che produca un campo magnetico rotante B1, ortogonale a B0 e oscillante ad onda continua lungo la direzione x. Quando, variando la frequenza dell’oscillatore, la frequenza di B1 entrerà in risonanza con le frequenze di Larmor, la magnetizzazione M0 sarà inclinata producendo una componente M nel piano xy, detta magnetizzazione trasversale, che può essere rivelata mediante una bobina posta sul piano xy.

78
Rilassamento Come la magnetizzazione ritorna all’equilibrio?
Come M0 ritorna lungo z? Come scompare o decade la magnetizzazione trasversale? Rilassamento Esistono due possibilità di rilassamento: longitudinale e trasversale Rilassamento longitudinale (T1): il ritorno da uno stato eccitato allo stato di equilibrio. Il rilassamento longitudinale può essere visto come ritorno alla distribuzione di Boltzmann tra i due stati di spin. T1 è la costante di tempo che descrive come la magnetizzazione trasversale ritorna al suo valore di equilibrio.
: il ritorno da uno stato eccitato allo stato di equilibrio. Il rilassamento longitudinale può essere visto come ritorno alla distribuzione di Boltzmann tra i due stati di spin. T1 è la costante di tempo che descrive come la magnetizzazione trasversale ritorna al suo valore di equilibrio.")
79
Rilassamento longitudinale (T1):
Il ritorno allo stato fondamentale può essere spontaneo o stimolato L’emissione spontanea è un fenomeno trascurabile per l’NMR poiché la DE tra i due stati è piccola. L’emissione stimolata richiede l’interazione del nucleo con un campo magnetico oscillante alla sua frequenza di risonanza. Le fonti di campi magnetici sono: Momenti magnetici di altri nuclei (principale meccanismo di rilassamento nelle molecole piccole e dipende da r-3) Momenti magnetici elettronici (per composti con elettroni spaiati) L’oscillazione è prodotta generalmente dal moto (rotazioni) termico della molecola
 Momenti magnetici elettronici (per composti con elettroni spaiati) L’oscillazione è prodotta generalmente dal moto (rotazioni) termico della molecola.")
80
Rilassamento trasversale (T2):
T2 (rilassamento spin-spin) è la costante di tempo che descrive il decadimento della magnetizzazione trasversale. Esso comporta il trasferimento di energia tra protoni in precessione la qual cosa produce una perdita di fase e di segnale. Un altro fattore importante che contribuisce alla perdita di segnale trasversale è la disomogeneità di campo.
 è la costante di tempo che descrive il decadimento della magnetizzazione trasversale. Esso comporta il trasferimento di energia tra protoni in precessione la qual cosa produce una perdita di fase e di segnale. Un altro fattore importante che contribuisce alla perdita di segnale trasversale è la disomogeneità di campo.")
81
Il tempo di rilassamento T2 regola anche la velocità di decadimento del FID in uno spettrometro ideale, in cui il campo magnetico è assolutamente omogeneo T2 t Rilassamento veloce (T2 piccolo) Rilassamento lento (T2 grande) Dal punto di vista microscopico, il rilassamento trasversale può essere visto come una perdita di fase dei nuclei che tuttavia non modificano il loro spin. Esso richiede solo interazioni tra nucleo e nucleo.
 Rilassamento lento. (T2 grande) Dal punto di vista microscopico, il rilassamento trasversale può essere visto come una perdita di fase dei nuclei che tuttavia non modificano il loro spin. Esso richiede solo interazioni tra nucleo e nucleo.")
82
Per molecole piccole che si muovono rapidamente in soluzioni non viscose il principale contributo a T2 è rappresentato dalla disomogeneità di campo. Per molecole piccole T1 e T2 sono molto simili. Per molecole grandi in soluzioni viscose T1>>T2 cioè il principale percorso di rilassamento è quello longitudinale

83
Esperimenti di doppia risonanza
Gli esperimenti di doppia risonanza prevedono l’uso di una bobina addizionale per irradiare un nucleo alla sua frequenza di risonanza ed osservare l’effetto determinato su un nucleo diverso ad una diversa frequenza di risonanza. Disaccoppiamento di spin Effetto Nucleare Overhauser (NOE).
.")
84
Disaccoppiamento di spin
Se un protone, in una molecola è irradiato con continuità, la radiazione elettromagnetica causa un continua variazione dello stato di spin di quel protone da a a b e viceversa. Le conseguenze dell’irradiazione continua sono: in seguito al continuo passaggio tra gli stati a e b, la differenza di popolazione tra i due stati si annulla. Per questo il segnale del nucleo irradiato sparisce dallo spettro. il nucleo accoppiato con quello irradiato non si trova più nelle vicinanze di un protone o nello stato a oppure nello stato b, ma nelle vicinanze di un protone che cambia continuamente il suo stato di spin per cui non risente più dell’accoppiamento e risuona come singoletto, cioè come se il nucleo irradiato non esistesse.

85
Questa informazione è di grande importanza nell’interpretazione di spettri di una certa complessità.

86
nI= I-I0 x 100 Io Effetto Nucleare Overhauser (NOE).
Il Noe può essere definito come la variazioni in intensità della risonanza di un nucleo quando le popolazioni dei due stati di spin di un altro nucleo sono perturbate rispetto al loro valore all’equilibrio. nI= I-I0 Io x 100 I due nuclei devono essere dipolarmente accoppiati, cioè sufficientemente vicini affinchè sia realizzabile, attraverso lo spazio, un interazione magnetica tra di essi.

87
L’effetto più pronunciato di questa interazione si riflette sul tempo di rilassamento T1 che per un dato nucleo dipende dall’interazioni con compi magnetici fluttuanti ad una frequenza corrispondente alla frequenza di risonanza del nucleo i questione.

88
Origine del Noe Consideriamo due nuclei con I=1/2, spazialmente vicini ma non scalarmente accoppiati. Saturiamo S e quindi forziamo il sistema in una situazione di non equilibrio con nuove popolazioni di spin Sistema all’equilibrio aa-ba=aa-ab=D ab-bb=ba-bb=D ab-ba=0 aa-bb=2D Sistema non all’equilibrio aa-ba=D ab-bb=D ab-ba=D aa-bb=D

89
Sistema non all’equilibrio ab-ba=D aa-bb=D Sistema all’equilibrio
Il sistema cercherà di ritornare nella condizione iniziale di popolazioni. Sistema non all’equilibrio ab-ba=D aa-bb=D Sistema all’equilibrio ab-ba=0 aa-bb=2D Attraverso processi di rilassamento incrociato

90
W2 e W0 competono l’un l’altro e il percorso dominante determina il segno del Noe. Inoltre le condizioni di equilibrio per I possono essere generate anche attraverso percorsi di rilassamento non incrociato, indipendenti da S (W1). Tutti i possibili cammini di rilassamento dipendono dalla distanza tra i due nuclei (inversamente proporzionale a r6) e dal tempo di correlazione rotazionale tc) tc: l’intervallo di tempo necessario alla molecola per ruotare di un angolo di 1 rad rispetto ad un asse tc= M x s
 e dal tempo di correlazione rotazionale tc) tc: l’intervallo di tempo necessario alla molecola per ruotare di un angolo di 1 rad rispetto ad un asse. tc= M x s.")
91
Noe positivo Noe negativo
Molecole piccole in soluzioni non viscose, che ruotano e si muovono velocemente, hanno piccoli valori di tc e rilassano preferenzialmente attraverso meccanismi W2 Noe positivo Molecole grandi che ruotano e si muovono lentamente hanno grandi valori di tc e rilassano preferenzialmente attraverso meccanismi W0 Noe negativo

92
Noe Difference

93
– = L'effetto nucleare Overhauser (NOE)
Poiché l'aumento di intensità è piccolo, normalmente il NOE viene misurato utilizzando spettri differenza: – Spettro con irradiazione = Spettro senza irradiazione NOE! Spettro differenza

94
Uso dell'effetto nucleare Overhauser
L’aumento della intensità del segnale di un protone in seguito all’irradiazione su un altro protone è in generale considerata prova della vicinanza spaziale (diciamo entro 3-4 Å) tra i due protoni. Non è vero il contrario, cioè non possiamo concludere dall’assenza di NOE che i due protoni sono lontani. Il NOE è di enorme utilità per la determinazione della stereochimica delle molecole. Per esempio se stiamo studiando la stereochimica di: un effetto NOE tra i protoni in 2 e in 4 indica vicinanza spaziale tra i due protoni, e questo è in accordo solo con il diastereoisomero cis.
 tra i due protoni. Non è vero il contrario, cioè non possiamo concludere dall’assenza di NOE che i due protoni sono lontani. Il NOE è di enorme utilità per la determinazione della stereochimica delle molecole. Per esempio se stiamo studiando la stereochimica di: un effetto NOE tra i protoni in 2 e in 4 indica vicinanza spaziale tra i due protoni, e questo è in accordo solo con il diastereoisomero cis.")
95
Il Noe in sistemi multi-spin
Per il nucleo I intervengono altri contributi al suo rilassamento data la vicinanza di N. In altre parole l’interazione dipolare tra I e N fa si che il rilassamento longitudinale di I non dipenda soltanto dall’influenza di S. In altre parole saturando S, l’effetto Noe prodotto su I è minore a causa della presenza di N

96
Il Noe transiente La velocità con cui il Noe cresce dipende soltanto dal rilassamento incrociato tra i due nuclei. Il modo più conveniente per ottenere questi dati cinetici è quello di invertire selettivamente la popolazione lungo le transizioni di un dato nucleo, aspettare un dato tempo variabile (tm) e poi acquisire lo spettro. La velocità di crescita del Noe ha una dipendenza lineare con il mixing time. Solo per bassi valori di tm (minori di T1) è valida la dipendenza dell’aumento del Noe in dipendenza esclusiva della distanza internucleare.
 e poi acquisire lo spettro. La velocità di crescita del Noe ha una dipendenza lineare con il mixing time. Solo per bassi valori di tm (minori di T1) è valida la dipendenza dell’aumento del Noe in dipendenza esclusiva della distanza internucleare.")
97
La distanza internucleare
Assumendo di realizzare esperimenti in cui il Noe cresce linearmente con il mixing time, l’intensità dell’effetto Noe registrato tra due spin A e B dopo un dato tm dipenderà da la distanza tra A e B e dal tc della molecola Poiché misure accurate del tempo di correlazione molecolare sono difficili è preferibile usare come riferimento l’effetto Noe tra due nuclei X e Y, che si trovano nella stessa molecola, la cui distanza è nota e paragonare l’intensità dell’effetto Noe con quello misurato tra due nuclei, A e B, per i quali la distanza è sconosciuta.

98
NOE e disaccoppiamento
Una irradiazione su un protone è in grado di provocare sia NOE che disaccoppiamento di spin, ed i due effetti avvengono contemporaneamente in un esperimento ad onda continua. Questo è un problema, perché il disaccoppiamento, facendo variare le molteplicità, rende difficile l'uso dell'esperimento differenza. In un esperimento a trasformata di Fourier, tuttavia, i due fenomeni possono essere tenuti ben distinti. Nel NOE la variazione dell’intensità del segnale è alla variazione della differenza tra le popolazioni degli stati a e b: l'intensità del segnale dipende dalla differenza di popolazione al momento dell'impulso, per cui è importante avere irradiazione prima dell’impulso, non dopo. Nel disaccoppiamento di spin l'irradiazione fa variare la frequenza a cui il nucleo emette, e quindi l'irradiazione deve avvenire durante l'emissione, cioè dopo l’impulso. Quindi: Irradiazione prima dell'impulso NOE Irradiazione dopo l'impulso disaccoppiamento







 Neutroni (n°) Elettroni (e) Gli atomi contengono diversi tipi di particelle subatomiche.>")









 tra la radiazione.>")


 Introduzione Disaccoppiamento di 1 H ed effetto NOE Intensità dei picchi.>")
>")