Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Farmaci antibatterici e resistenza batterica ai chemioterapici

2
La scoperta della penicillina
Fleming studiando varianti dello stafilococco, osservò che una muffa che contaminava una delle sue culture aveva inibito intorno a sé la crescita dello stafilococco osservò che il brodo di coltura in cui erano cresciuti i funghi possedeva un potente effetto inibitorio nei confronti di molti microrganismi. Chain e Florey Identificarono il principio attivo - PENICILLINA - e la sintetizzarono

3
P.Herlich Domakg Trèfoul
“proiettile magico” e teoria della “tossicità selettiva” SALVARSAN è una arsfenamina attiva selettivamente verso Treponema pallidum Salvarsan è metabolizzato in vivo al composto arsenossido Domakg riprese il concetto di tossicità selettiva PRONTOSIL: capostipite dei sulfamidici Trèfoul dimostrazione che prontosil è trasformato in vivo a sulfanilamide

4
Definizione e caratteristiche
Antibiotici: sostanze prodotte da varie specie di microrganismi (batteri o funghi) che sopprimono la crescita di altri microrganismi e ne possono causare la distruzione Chemioterapici: farmaci antibatterici prodotti per sintesi chimica Farmaco batteriostatico: ferma la moltiplicazione batterica, permettendo alle difese dell’ospite di reagire, ma non uccide le cellule Farmaco battericida: uccide i microrganismi
 che sopprimono la crescita di altri microrganismi e ne possono causare la distruzione. Chemioterapici: farmaci antibatterici prodotti per sintesi chimica. Farmaco batteriostatico: ferma la moltiplicazione batterica, permettendo alle difese dell’ospite di reagire, ma non uccide le cellule. Farmaco battericida: uccide i microrganismi.")
5
Selettività Idealmente, una sostanza ad azione antimicrobica dovrebbe avere effetto massimo sulla cellula batterica e scarso o nessun effetto sulle cellule umane Alcune attività metaboliche della cellula batterica differiscono significativamente da quelle delle cellule umane Le molecole ad attività antibiotica o chemioterapica sfruttano queste differenze Tutti i farmaci antibatterici agiscono inibendo una determinata via metabolica, per cui sono attivi solo nei confronti di batteri attivamente metabolizzanti

6
Fonti di antibatterici
MICROORGANISMI : bacitracina e polimixina sono prodotte da batteri appartenenti al genere Bacillus, streptomicina e tetraciclina dal genere streptomyces, gentamicina da micromospora purpurea, la griseofulvina, alcune penicilline e le cefalosporine da diversi miceti (Penicillium e Acremonium). La maggior parte degli antibiotici utilizzati deriva da batteri del genere Streptomyces SINTESI SEMISINTESI: parte della molecola è prodotta con un processo fermentativo utilizzando un microrganismo; successivamente la molecola viene modificata mediante un processo chimico. Penicilline e cefalosporine
. La maggior parte degli antibiotici utilizzati deriva da batteri del genere Streptomyces. SINTESI. SEMISINTESI: parte della molecola è prodotta con un processo fermentativo utilizzando un microrganismo; successivamente la molecola viene modificata mediante un processo chimico. Penicilline e cefalosporine.")
7
Bersagli degli antibiotici
Parete cellulare Sintesi proteica DNA e RNA Metaboliti In fatti ……..

8
inibizione della sintesi degli acidi nucleici
Inibizione della parete cellulare attività anti-metabolica inibizione della sintesi degli acidi nucleici inibizione della sintesi delle proteine alterazione delle membrane cellulari

9
Antibiotici che agiscono sulla sintesi della parete cellulare
Il 90-95% è costituita da mureina (o peptidoglicano). acidi teicoici acidi teicuronici proteine (molto rare) La membrana interna o citoplasmatica: doppio strato di fosfolipidi (lo strato rigido è costituito da un reticolo di peptidoglicani La membrana esterna ha una complessa impalcatura di molecole che si protendono all'esterno (a modo di "sensori") e di proteine (tra cui le porine) che permettono il passaggio di molecole o cariche ioniche tra l'interno e l'esterno della cellula
. acidi teicoici. acidi teicuronici. proteine (molto rare) La membrana interna o citoplasmatica: doppio strato di fosfolipidi (lo strato rigido è costituito da un reticolo di peptidoglicani. La membrana esterna ha una complessa impalcatura di molecole che si protendono all esterno (a modo di sensori ) e di proteine (tra cui le porine) che permettono il passaggio di molecole o cariche ioniche tra l interno e l esterno della cellula.")
10
PEPTIDOGLICANO. STRUTTURA e SINTESI
Model of peptidoglycan synthesis. Precursors are produced in the cytoplasm subsequently coupled to a undecaprenyl monophosphate, the carrier lipid, and modified. After translocation to the outer face, peptidoglycan is assembled and incorporated into the cell wall. MraY, UDP-MurNAc-pentapeptide phosphotransferase; MurA, UDP-GlcNAc enolpyruvyl transferase; MurB, UDP-MurNAc dehydrogenase; MurC, UDP-MurNAc–L-Ala ligase; MurD, UDP-MurNAc-L-Ala–D-Glu ligase; MurE, UDP-MurNAc-L-Ala-D-Glu–meso-Dap ligase; MurF, UDP-MurNAc-tripeptide–D-alanyl-D-Ala ligase; MurG, UDP-GlcNAc-undecaprenoyl-pyrophosphoryl-MurNAc-pentapeptide transferase; MurI, Glu racemase; PEP, phosphoenol

11
Antibiotici attivi sulla prima tappa della sintesi del peptidoglicano
La prima tappa avviene nel citoplasma, dove vengono sintetizzate le unità fondamentali della struttura portante del peptidoglicano NAG e NAM La D-CICLOSERINA è un analogo strutturale della D-Alanina. Essa inibisce in maniera competitiva due enzimi coinvolti nella formazione del dipeptide D-Alanil-D-alanina: racemasi che converte L-alanina nel suo stereoisomero D, e sintetasi che catalizza la formazione del legame peptidico tra le due molecole D-alanina La cicloserina è neurotossica e non viene usata clinicamente, eccetto per il trattamento delle infezioni da Mycobacterium tuberculosis resistenti agli altri farmaci

12
Fosfomicina La FOSFOMICINA è un analogo strutturale del fosfo-enolpiruvato. La reazione di condensazione tra UDP-N-acetil glucosamina e fosfoenolpiruvato negli stadi iniziali della sintesi del peptidoglicano rappresenta il target d’azione della fosfomicina Impedisce la sintesi dell’acido N-acetil-muramico, legandosi covalentemente al sito attivo dell’enzima piruvato-UDP-NAG trasferasi La rapida insorgenza di resistenza la rendono praticamente inutile in clinica

13
Antibiotici attivi sulla seconda tappa della sintesi del peptidoglicano
La seconda tappa della sintesi del peptidoglicano avviene sulla superficie interna della membrana citoplasmatica dove l’ N-acetilmuramilpeptide viene legato da un trasportatore lipidico che trasloca la subunità completa all’esterno della membrana citoplasmatica La BACITRACINA inibisce la defosforilazione del trasportatore lipidico legandosi al bactoprenolo-difosfato e bloccando la rigenerazione del bactoprenolo-monofosfato. E’ un polipeptide ciclico tossico per l’uso clinico umano Viene usato per trattamenti topici e come additivo alimentare per ruminanti al fine di ridurre la produzione di metanolo nel rumine

14
Antibiotici attivi sulla terza tappa della sintesi del peptidoglicano
La terza tappa avviene nel contesto del peptidoglicano e nello spazio periplasmico (Gram-), dove l’unità basale, liberata dal legame del trasportatore lipidico, si unisce all’ estremità in accrescimento di una catena di peptidoglicano (transpeptidazione). Molti antibiotici agiscono su questa tappa. .
, dove l’unità basale, liberata dal legame del trasportatore lipidico, si unisce all’ estremità in accrescimento di una catena di peptidoglicano (transpeptidazione). Molti antibiotici agiscono su questa tappa. .")
15
La vancomicina è un farmaco antibiotico prodotto da Streptococcus orientalis che fa parte, insieme alla teicoplanina, della classe dei glicopeptidi. Sono molecole ad alto peso molecolare, che agiscono inibendo la polimerizzazione della parete del peptidoglicano dei batteri gram positivi. L'alto peso molecolare non consente a tali molecole l'attraversamento della membrana cellulare esterna dei germi Gram negativi, per cui la vancomicina è inefficace contro questi batteri; unica eccezione sono i Flavobacterium. Essendo stata diffusamente impiegata per debellare infezioni da Staphylococcus aureus si sono sviluppate resistenze, fino alla nascita di ceppi di S. aureus particolarmente resistenti (VISA: Vancomicine intermediate-resistant S. aureus). La VANCOMICINA e la RISTOCETINA sono glicopeptidi che si legano all’estremità D-alanina-D-alanina del pentapeptide legato al bactoprenolo ed impediscono l’azione della transpeptidasi sequestrandone il substrato. La RISTOCETINA non è più utilizzata in clinica per la sua tossicità. Provoca agglutinamento di placchette e coagulazione sanguigna. Si usa negli saggi di queste funzioni in vitro

16
Un’altro analogo della vancomicina d’uso clinico:TEICOPLANINA

17
Antibiotici attivi sulla terza tappa del metabolismo del peptidoglicano: gli antibiotici β-lattamici
Tutte le operazioni terminali di polimerizzazione, transpeptidazione ed inserimento delle unità peptidoglicaniche nella parete cellulare sono catalizzate da una serie di enzimi che presentano la caratteristica assolutamente peculiare di legare covalentemente la penicillina ed altri antibiotici β-lattamici e sono noti, pertanto, come ”proteine leganti la penicillina” o PBP (Penicillin-binding proteins) Transpeptidasi (TPD): formazione legami peptidici (Integrità strutturale) Endopeptidasi (EPD): rompe legami peptidici formati da TPD, consentendo l’allungamento del peptidoglicano nella divisione cellulare (mantenimento della forma) Carbossipeptidasi (CPD): rimuove una D-Ala terminale dal dimero bloccando la TPD; regola la formazione dei legami peptidici (divisione cellulare)
 Transpeptidasi (TPD): formazione legami peptidici (Integrità strutturale) Endopeptidasi (EPD): rompe legami peptidici formati da TPD, consentendo l’allungamento del peptidoglicano nella divisione cellulare. (mantenimento della forma) Carbossipeptidasi (CPD): rimuove una D-Ala terminale dal dimero bloccando la TPD; regola la formazione dei legami peptidici (divisione cellulare)")
18
Meccanismo d’azione Gli antibiotici β-lattamici sono analoghi strutturali del dipeptide D-alanina-D-alanina che devono la loro azione antibatterica proprio alla capacità di legarsi a questi enzimi inibendo la sintesi del peptidoglicano representation of the mode of action of β-lactams and glycopeptides and their chemical structures. Penicillin-binding proteins (PBPs), enzymes that catalyze the last steps of bacterial cell wall biosynthesis, bind to the terminal D-alanyl-D-alanine residues of peptidoglycan precursors to carry out transpeptidation (A). β-lactam antibiotics (β) are structurally analogous to D-Ala-D-Ala. PBPs can react with these antibiotics by cleaving the β-lactam bond and forming a stable intermediate that does not react further. Acylation of PBPs by β-lactams therefore inactivates the enzyme (B). Glycopeptides (G) inhibit cell wall synthesis by a completely different mechanism. They bind the terminal D-Ala-D-Ala residues of the peptidoglycan precursor preventing access of PBPs to their natural substrate (C).
, enzymes that catalyze the last steps of bacterial cell wall biosynthesis, bind to the terminal D-alanyl-D-alanine residues of peptidoglycan precursors to carry out transpeptidation (A). β-lactam antibiotics (β) are structurally analogous to D-Ala-D-Ala. PBPs can react with these antibiotics by cleaving the β-lactam bond and forming a stable intermediate that does not react further. Acylation of PBPs by β-lactams therefore inactivates the enzyme (B). Glycopeptides (G) inhibit cell wall synthesis by a completely different mechanism. They bind the terminal D-Ala-D-Ala residues of the peptidoglycan precursor preventing access of PBPs to their natural substrate (C).")
19
A differente struttura molecolare corrisponde un diverso antibiotico
Gli antibiotici β-lattamici Gli antibiotici b-lattamici sono accomunati dalla presenza di un anello tetratomico azetidinico b-lattamico Penicilline Penam (tiazolidine) A differente struttura molecolare corrisponde un diverso antibiotico b-lattami β-Lactams containing thiazolidine rings are named penams. β-Lactams containing pyrrolidine rings are named carbapenams. β-Lactams fused to oxazolidine rings are named oxapenams or clavams. β-Lactams fused to unsaturated five-membered rings: β-Lactams containing 2,3-dihydrothiazole rings are named penems. β-Lactams containing 2,3-dihydro-1H-pyrrole rings are named carbapenems. β-Lactams fused to unsaturated six-membered rings: β-Lactams containing 3,6-dihydro-2H-1,3-thiazine rings are named cephems. β-Lactams containing 1,2,3,4-tetrahydropyridine rings are named carbacephems. β-Lactams containing 3,6-dihydro-2H-1,3-oxazine rings are named oxacephems. β-Lactams not fused to any other ring are named monobactams. The cephalosporins (sg. pronounced /ˌsɛfəlɵˈspɔrɨn/) are a class of β-lactam antibiotics originally derived from Acremonium, which was previously known as "Cephalosporium".[1] Together with cephamycins they constitute a subgroup of β-lactam antibiotics called cephems Cefalosporine Cephem (diidrotiazine)
 A differente struttura molecolare corrisponde un diverso antibiotico. b-lattami. β-Lactams containing thiazolidine rings are named penams. β-Lactams containing pyrrolidine rings are named carbapenams. β-Lactams fused to oxazolidine rings are named oxapenams or clavams. β-Lactams fused to unsaturated five-membered rings: β-Lactams containing 2,3-dihydrothiazole rings are named penems. β-Lactams containing 2,3-dihydro-1H-pyrrole rings are named carbapenems. β-Lactams fused to unsaturated six-membered rings: β-Lactams containing 3,6-dihydro-2H-1,3-thiazine rings are named cephems. β-Lactams containing 1,2,3,4-tetrahydropyridine rings are named carbacephems. β-Lactams containing 3,6-dihydro-2H-1,3-oxazine rings are named oxacephems. β-Lactams not fused to any other ring are named monobactams. The cephalosporins (sg. pronounced /ˌsɛfəlɵˈspɔrɨn/) are a class of β-lactam antibiotics originally derived from Acremonium, which was previously known as Cephalosporium .[1] Together with cephamycins they constitute a subgroup of β-lactam antibiotics called cephems. Cefalosporine. Cephem. (diidrotiazine)")
20
Le penicilline sono attive contro la maggior parte dei batteri gram positivi come gli stafilococchi e gli streptococchi, contro gli spirilli (Treponema pallidum), contro gonococchi e meningococchi. Nonostante le penicilline siano antibiotici battericidi, a causa dell'intenso uso che se ne è fatto ed alla produzione di particolari enzimi (penicillinasi o β-lattamasi ) da parte dei germi, spesso si incorre in fenomeni di resistenza batterica. Le penicilline naturali sono inattive nei confronti del batterio della tubercolosi e dei germi gram negativi. Le penicilline semi-sintetiche, prodotte da colture biologiche di Penicillium, hanno in comune un sistema beta-lattamico-diazolico e hanno un ampio spettro di azione, anche contro alcuni batteri gram negativi.
 da parte dei germi, spesso si incorre in fenomeni di resistenza batterica. Le penicilline naturali sono inattive nei confronti del batterio della tubercolosi e dei germi gram negativi. Le penicilline semi-sintetiche, prodotte da colture biologiche di Penicillium, hanno in comune un sistema beta-lattamico-diazolico e hanno un ampio spettro di azione, anche contro alcuni batteri gram negativi..")
21
Altre penicilline

22
Alcune cefalosporine

23
Altre b-lattamici Imipenem Carbapenem Carbapenam (diidropirrole)
(pirrolidine) Meropenem β-Lactams containing pyrrolidine rings are named carbapenams. β-Lactams fused to oxazolidine rings are named oxapenams or clavamsβ-Lactams containing 2,3-dihydrothiazole rings are named penems. β-Lactams containing 2,3-dihydro-1H-pyrrole rings are named carbapenems. β-Lactams fused to unsaturated six-membered rings: β-Lactams containing 3,6-dihydro-2H-1,3-thiazine rings are named cephems. β-Lactams containing 1,2,3,4-tetrahydropyridine rings are named carbacephems. β-Lactams containing 3,6-dihydro-2H-1,3-oxazine rings are named oxacephems. β-Lactams not fused to any other ring are named monobactams. The cephalosporins (sg. pronounced /ˌsɛfəlɵˈspɔrɨn/) are a class of β-lactam antibiotics originally derived from Acremonium, which was previously known as "Cephalosporium".[1] Together with cephamycins they constitute a subgroup of β-lactam antibiotics called cephems Penem (diidrotiazole) Faropenem Carbacephem (tetraidropiridine) Sulbactam
 Meropenem. β-Lactams containing pyrrolidine rings are named carbapenams. β-Lactams fused to oxazolidine rings are named oxapenams or clavamsβ-Lactams containing 2,3-dihydrothiazole rings are named penems. β-Lactams containing 2,3-dihydro-1H-pyrrole rings are named carbapenems. β-Lactams fused to unsaturated six-membered rings: β-Lactams containing 3,6-dihydro-2H-1,3-thiazine rings are named cephems. β-Lactams containing 1,2,3,4-tetrahydropyridine rings are named carbacephems. β-Lactams containing 3,6-dihydro-2H-1,3-oxazine rings are named oxacephems. β-Lactams not fused to any other ring are named monobactams. The cephalosporins (sg. pronounced /ˌsɛfəlɵˈspɔrɨn/) are a class of β-lactam antibiotics originally derived from Acremonium, which was previously known as Cephalosporium .[1] Together with cephamycins they constitute a subgroup of β-lactam antibiotics called cephems. Penem. (diidrotiazole) Faropenem. Carbacephem. (tetraidropiridine) Sulbactam.")
24
Monobactams Oxacephem Clavam Moxalactam (ossazolidine) Clavulanico
Monobactams are beta-lactam compounds wherein the beta-lactam ring is alone and not fused to another ring (in contrast to most other beta-lactams, which have at least two rings). They work only against Gram-negative bacteria.[citation needed] The only commercially available monobactam antibiotic is aztreonam. Other examples of monobactams are tigemonam,[1] nocardicin A, and tabtoxin. Monobactam is a monocyclic beta-lactam antibiotic (a monobactam) originally isolated from Chromobacterium violaceum. Monobactam exhibits potent and specific activity in vitro against a wide spectrum of gram-negative aerobic pathogens including Pseudomonas aeruginosa. It has no useful activity against gram-positive bacteria or anaerobes, but has very broad spectrum against gram-negative aerobes, including Pseudomonas aeruginosa. This has given it the nickname "the magic bullet for aerobic gram-negative bacteria". Monobactam, unlike the majority of beta-lactam antibiotics, does not induce beta-lactamase activity and its molecular structure confers a high degree of resistance to hydrolysis by beta-lactamases (such as penicillinases and cephalosporinases) produced by most gram-negative and gram-positive pathogens; it is, therefore, usually active against gram-negative aerobic microorganisms that are resistant to antibiotics hydrolyzed by beta-lactamases. It is active against many strains that are multiply-resistant to other antibiotics, such as certain cephalosporins, penicillin, and aminoglycosides. Monobactam maintains its antimicrobial activity over a pH range of 6 to 8 in vitro, as well as in the presence of human serum and under anaerobic conditions Aztreonam Tigemonam
. They work only against Gram-negative bacteria.[citation needed] The only commercially available monobactam antibiotic is aztreonam. Other examples of monobactams are tigemonam,[1] nocardicin A, and tabtoxin. Monobactam is a monocyclic beta-lactam antibiotic (a monobactam) originally isolated from Chromobacterium violaceum. Monobactam exhibits potent and specific activity in vitro against a wide spectrum of gram-negative aerobic pathogens including Pseudomonas aeruginosa. It has no useful activity against gram-positive bacteria or anaerobes, but has very broad spectrum against gram-negative aerobes, including Pseudomonas aeruginosa. This has given it the nickname the magic bullet for aerobic gram-negative bacteria . Monobactam, unlike the majority of beta-lactam antibiotics, does not induce beta-lactamase activity and its molecular structure confers a high degree of resistance to hydrolysis by beta-lactamases (such as penicillinases and cephalosporinases) produced by most gram-negative and gram-positive pathogens; it is, therefore, usually active against gram-negative aerobic microorganisms that are resistant to antibiotics hydrolyzed by beta-lactamases. It is active against many strains that are multiply-resistant to other antibiotics, such as certain cephalosporins, penicillin, and aminoglycosides. Monobactam maintains its antimicrobial activity over a pH range of 6 to 8 in vitro, as well as in the presence of human serum and under anaerobic conditions. Aztreonam. Tigemonam.")
25
Meccanismi chimici ed enzimatici di degradazione delle penicilline
L’anello β-lattamico è il punto debole degli antibiotici β-lattamici e il legame CO-N è idrolizzato da alcuni enzimi (β-lattamasi) o in medio acido con conseguente produzione di acido penicilloico inattivo
 o in medio acido con conseguente produzione di acido penicilloico inattivo.")
26
β-lattamasi Sono le principali responsabile della insorgenza di resistenza I diversi microrganismi elaborano distinte β-lattamasi, anche se la maggior parte dei batteri produce una sola forma dell’enzima La specificità di substrato di alcuni di questi enzimi è relativamente ristretta, per questo motivo alcuni di questi enzimi sono spesso definiti penicillinasi o cefalosporinasi; altri enzimi ad ampio spettro d’azione sono meno discriminanti e possono idrolizzare differenti antibiotici β-lattamici Modello strutturale delle β-lattamasi. L’esame cristallografico suggerisce un ruolo importante del residuo 244. L’arginina che occupa questo sito sembra essere critica per il legame agli antibiotici e per la capacità degli inibitori suicidi di inattivare l’enzima

27
Alcune molecole sono in grado di legarsi alle β-lattamasi inattivandole e prevenendo, così la distruzione degli antibiotici β-lattamici. L’acido clavulanico ed il sulbactam hanno una debole attività antimicrobica intrinseca, ma sono inibitori suicidi (formano un legame irreversibile) delle β-lattamasi ACIDO CLAVULANICO SULBACTAM
 delle β-lattamasi. ACIDO CLAVULANICO. SULBACTAM.")
28
Acido Clavulanico-Meccanismo di inibizione delle -Lattamasi

29
Penicillina acido-labile, resistente alle β-lattamasi:
Penicilline ad ampio spettro, adatte alla somministrazione orale (resistenti ad acidi): Penicillina acido-labile, resistente alle β-lattamasi: AMPICILLINA AMOXICILLINA L'amoxicillina è un antibiotico appartenente alla classe dei β-lattamici al gruppo delle penicilline semisintetiche, viene usata da sola ed in associazione con l'acido clavulanico come antibiotico in molte infezioni sostenute da germi sensibili, che colpiscono l'orecchio, la gola, le vie respiratorie inferiori, la cute e l'apparato urinario. Recentemente viene spesso usata insieme con altri antibiotici (claritromicina, ecc) ed inibitori di pompa protonica (omeprazolo, esomeprazolo, ecc) per l'eradicazione dell'Helicobacter pylori germe molto resistente al pH acido che causa un'infezione cronica dello stomaco, la gastrite, a sua volta concausa di svariate complicazioni spesso anche fatali (Ulcera gastrica e cancro dello stomaco). La meticillina per via della sua alta tossicità non ha trovato l'utilizzo come farmaco da somministrare durante infezioni, ma ugualmente viene utilizzato per esperimenti METICILLINA
: Penicillina acido-labile, resistente alle β-lattamasi: AMPICILLINA. AMOXICILLINA. L amoxicillina è un antibiotico appartenente alla classe dei β-lattamici al gruppo delle penicilline semisintetiche, viene usata da sola ed in associazione con l acido clavulanico come antibiotico in molte infezioni sostenute da germi sensibili, che colpiscono l orecchio, la gola, le vie respiratorie inferiori, la cute e l apparato urinario. Recentemente viene spesso usata insieme con altri antibiotici (claritromicina, ecc) ed inibitori di pompa protonica (omeprazolo, esomeprazolo, ecc) per l eradicazione dell Helicobacter pylori germe molto resistente al pH acido che causa un infezione cronica dello stomaco, la gastrite, a sua volta concausa di svariate complicazioni spesso anche fatali (Ulcera gastrica e cancro dello stomaco). La meticillina per via della sua alta tossicità non ha trovato l utilizzo come farmaco da somministrare durante infezioni, ma ugualmente viene utilizzato per esperimenti. METICILLINA.")
30
Penicilline ß-lattamasi-resistenti, adatte alla somministrazione orale:
CLOXACILLINA OXACILLINA L'oxacillina è efficace contro gli enzimi penicillinasi prodotti da alcuni batteri, quali Staphylococcus aureus. Tuttavia sono stati identificati ceppi resistenti, inseribili nella classe Oxacillin-resistant Staphylococcus aureus (ORSA). DICLOXACILLINA
. DICLOXACILLINA.")
31
Antibiotici che agiscono sulla membrana citoplasmatica: le polimixine
Le polimixine sono un gruppo di antibiotici attivi solo nei confronti dei batteri Gram-negativi alla cui membrana esterna si legano specificamente distruggendone le proprietà osmotiche e provocando la fuoriuscita di composti intracellulari In conseguenza del loro meccanismo d’azione le polimixine sono piuttosto tossiche anche per le cellule eucariotiche e il loro impiego è limitato ai trattamenti topici

32
Struttura chimica di una polimixina
Le polimixine sono molecole costituite da un peptide ciclico, legato a un polipeptide lineare che termina con una molecola di acido grasso. La presenza nella molecola di una porzione idrofila e una idrofoba consente a questi antibiotici di inserirsi tra lo strato proteico e quello lipidico alterando la permeabilità della membrana.

33
Il loro meccanismo d’azione è abbastanza peculiare in quanto sono antibiotici che agiscono in maniera analoga ai disinfettanti, provocando, cioè, l’alterazione di strutture cellulari piuttosto che inibendo processi biosintetici. Esse, pertanto, sono attive anche nei confronti di batteri non metabolizzanti. Le polimixine sono detergenti cationici che agiscono rimpiazzando competitivamente gli ioni Mg2+ e Ca2+ dei gruppi fosfato carichi negativamente delle membrane lipidiche. Il risultato è la distruzione delle membrane

34
Antibiotici attivi sulla sintesi proteica
Un gruppo numeroso di antibiotici deve la propria azione antibatterica all’interferenza con la sintesi proteica intervenendo con diversi meccanismi di cui i principali sono rappresentati dalla interazione con l’una o l’altra delle subunità ribosomiali 30S e 50S Poiché i ribosomi batterici sono significativamente diversi dai ribosomi delle cellule eucariotiche, tutti gli antibiotici che interagiscono con le subunità ribosomiali sono abbastanza selettivi per poter essere impiegati nella terapia antibatterica

35
Antibiotici che agiscono sulle subunità ribosomiale

36
Aminoglicosidi Gli aminoglicosidi (streptomicina, gentamicina,etc.) consistono di due o più aminozuccheri uniti da un legame glicosidico a un nucleo esoso che generalmente è in posizione centrale. Sono antibiotici attivi nei confronti di batteri sia Gram-positivi che Gram-negativi. Mentre la maggior parte degli inibitori della sintesi proteica microbica è batteriostatica, gli aminoglicosidi sono battericidi. Questa classe di antibiotici è oggi poco utilizzata, sia perché causa di numerosi effetti collaterali che per la frequente comparsa di resistenza batterica
 consistono di due o più aminozuccheri uniti da un legame glicosidico a un nucleo esoso che generalmente è in posizione centrale. Sono antibiotici attivi nei confronti di batteri sia Gram-positivi che Gram-negativi. Mentre la maggior parte degli inibitori della sintesi proteica microbica è batteriostatica, gli aminoglicosidi sono battericidi. Questa classe di antibiotici è oggi poco utilizzata, sia perché causa di numerosi effetti collaterali che per la frequente comparsa di resistenza batterica.")
37
. Complesso gentamicina-RNA target STREPTOMICINA GENTAMICINA
Structure of the antibiotic gentamicin C1a bound to its RNA target. The structure was determined using heteronuclear NMR spectroscopy. Gentamicin, an aminoglycoside antibiotic, binds within the major groove of the RNA, which is located in the decoding site of the bacterial ribosome. Aminoglycoside antibiotics cause misreading of the genetic code. Binding of the drug causes a conformational change in ribosomal RNA that dissrupts high-fidelity reading of the genetic code. Structure of aminoglycoside-ribosomal RNA complexes explain antibiotic resistance mechanisms. 3D structure of the aminoglycoside paromomycin bound to the bacterial rRNA decoding site. The sites of chemical modifications to either the antibiotic or RNA that lead to resistance are highlighted with purple spheres. These are clustered at the drug-RNA interface, such that steric or electrostatic penalties to drug-RNA affinity are introduced. The N7 methylation at G1405 only causes resistance to aminoglycosides like gentamicin that contact this position directly. The ribosome is the target of many clinically important antibiotics, such as aminoglycosides, tetracyclines, macrolides, streptogramins and oxazolidinones. We have previously focused on the aminoglycosides as a paradigm for ribosome- and RNA-directed antibiotics. Aminoglycosides bind to a conserved domain of 16S rRNA near the site of codon-anticodon interaction, and cause misreading of the genetic code. Our structural and mechanistic work revealed the structural basis for drug-RNA recogntion, the origins of antibiotic resistance and the molecular basis for toxicity. We are currently using the single-molecule fluorescence approach to understand the effect of drug binding on ribosome dynamics. Our goals are to: 1. Determine the effects of drugs on ligand and domain movements 2. Determine the mechanism of action for drugs whose mode of action is currently unclear. • The mechanism of eukaryotic translation initiation and elongation. Eukaryotic translation occurs by the same basic mechanism as in prokaryotic organisms. However, the processes of initiation and elongation are more complex and regulated. We are using a number of biophysical and structural approaches to characterized eukaryotic translation. These include: 1. Single-molecule fluorescence spectroscopy (with Prof. Steve Chu, Applied Physics, Stanford) to characterized the mechanism of translation initiation in yeast. 2. Single-molecule fluorescence analysis of the mechanism of translation elongation in yeast. 3. NMR structural studies of translation initiation factors, and proteins involved in cell signaling with translation as an endpoint (eg. PKR system). • The structure and function of the Hepatitis C virus (HCV) internal ribosome entry site. Figure 9 HCV polyprotein is translated using an internal ribosome entry site. Translation of most eukaryotic mRNA occurs in a 5âcap-dependent fashion. A number of initiation factors are involved in eventual recruitment of the 40S subunit, and subsequent ATP-dependent scanning to the first start codon. In contrast, translation of HCV RNA occurs in a cap-independent manner; the 40S subunit binds directly to the IRES, and only a subset of the initiation factors are required. Figure 10 Proteins mediate the IRES-40S subunit interaction. Our lab has identified a number of human ribosomal proteins that mediate IRES interaction with the 40S subunit. The positions of these proteins that have bacterial homologs is shown on the 30S ribosome crystal structure (left). These proteins are located on the protein-rich backside of the 40S subunit, and agree with the low resolution cryoEM study of the IRES-40S subunit solved bythe Frank and Doudna groups (shown on the right). Figure 11 Structure of a domain of the HCV IRES RNA solved by NMR. The secondary structure of the 100 kDa IRES is shown, with domain IIb highlighted. The RNA adopts a loop E motif, with an S-turn in the backbone, as shown in the superposition of final NMR structures. HCV is an important health problem, and infects more the 2% of the worlds population. HCV is an positive-strand RNA virus, and its RNA genome is translated immediately after uncoating of the virus in the cytoplasm of infected (liver) cells. An internal ribosome entry site (IRES) mediates the initiation of translation of the HCV genome. The 40S ribosomal subunit binds directly to the IRES RNA structure, and thus avoids the need for a 5â cap and scanning; upon IRES binding, the start codon for translation is positioned near the P site. We have been studying the mechanism and structure of the HCV IRES. We have previously delineated the domains of the IRES that are critical for 40S subunit binding, and the proteins on the surface of the 40S subunit that mediate this interaction. Our current interests include: 1. Determination of the structure of the IRES using NMR spectroscopy. The IRES is a 100 kDa RNA, and we are using novel NMR approaches, including residual dipolar couplings. 2. High resolution NMR structures of isolated IRES RNA domains. 3. Determining the conformational changes that the IRES undergoes on 40S subunit binding. 4. Understanding the mechanism of IRES-mediated initiation. What are the order of bidning events? How do initiation factors influence the process? What is the rate of initiation? How does IRES conformation affect initiation rate? • RNA-protein interactions in HIV virus. Figure 12 The Tat-TAR protein RNA complex regulates transcription of HIV and BIV RNAs. The TAR element is located at the 5â end of both retroviral RNAs. In HIV, the Tat protein interacts with a trinucleotide bulge, and a host cyclin interacts with a hairpin loop. In BIV, the Tat protein binds with high affinity to the RNA site, and cyclin does not interact with the hairpin looo. (right) Three-dimensional structure of the HIV Tat-TAR interaction solved by NMR. Superposition of final NMR structures of the BIV Tat-peptide-TAR RNA complex solved using 13C, 15N-labeled BIV peptide. The peptide forms a beta turn within the RNA major groove. Figure 13 A host tRNALys3 primes reverse transcription of HIV genomic RNA. A binary complex of tRNA and genomic RNA is specifically recognized by HIV reverse transcriptase. We are studying the structure of the RNA-RNA complex using NMR. Figure 14 HIV genomic RNA and tRNA form a high-affinity complex. Imino proton NMR spectrum of free tRNALys3 and a 1:1 complex (50 kDa total) with HIV genomic RNA. RNA plays a criticial role in the HIV virus replication cycle. We have a longstanding interest in critical protein-RNA interactions that occur in HIV biology. First, the Tat protein is a transcriptional regulator coded by the HIV virus. It binds to a stem-loop structure at the 5â-end of HIV mRNAs, and binding to the nascent transcript stimulates transcription. We have studied the HIV and Bovine immunodeficiency virus (BIV) Tat-TAR interaction using peptides that correspond to the RNA-binding domains. We have refined the structure of the BIV Tat-TAR interaction using modern heteronuclear NMR methods. The protein adopts a beta-turn conformation upon RNA binding. We are using this as a template to design small molecule inhibitors of the Tat-TAR interaction. Current interests include: 1. Design of cyclic peptide mimics of the BIV Tat peptide that recognized TAR RNA. 2. Design of cyclic peptides that interact with HIV TAR RNA. A complex between a human tRNALys and HIV genomic RNA is the priming site for reverse transcription of the HIV RNA genome. HIV reverse transcriptase (RT) specifically recognizes this RNA-RNA complex. We are investigating the structural basis for initiation of RT using a combination of methods. The results of this work may have important ramifications for the mechanism of action of RT inhibitors. Our current work focuses on: 1. NMR structure determination of the 50 kDa tRNALys3-genomic RNA binary complex. 2. x-ray crystallographic analys of the RT initiation complex 3. The use of NMR to study the RT initiation complex. Joseph Puglisi Professor and Chair, Department of Structural Biology Director, Stanford Magnetic Resonance Laboratory ph fx Mailing Address: Stanford University School of Medicine D105 Fairchild Science Building 299 Campus Drive West Stanford, CA Manolia Margaris Administrative Associate ph fx GENTAMICINA Complesso gentamicina-RNA target
 to characterized the mechanism of translation initiation in yeast. 2. Single-molecule fluorescence analysis of the mechanism of translation elongation in yeast. 3. NMR structural studies of translation initiation factors, and proteins involved in cell signaling with translation as an endpoint (eg. PKR system). • The structure and function of the Hepatitis C virus (HCV) internal ribosome entry site. Figure 9 HCV polyprotein is translated using an internal ribosome entry site. Translation of most eukaryotic mRNA occurs in a 5âcap-dependent fashion. A number of initiation factors are involved in eventual recruitment of the 40S subunit, and subsequent ATP-dependent scanning to the first start codon. In contrast, translation of HCV RNA occurs in a cap-independent manner; the 40S subunit binds directly to the IRES, and only a subset of the initiation factors are required. Figure 10 Proteins mediate the IRES-40S subunit interaction. Our lab has identified a number of human ribosomal proteins that mediate IRES interaction with the 40S subunit. The positions of these proteins that have bacterial homologs is shown on the 30S ribosome crystal structure (left). These proteins are located on the protein-rich backside of the 40S subunit, and agree with the low resolution cryoEM study of the IRES-40S subunit solved bythe Frank and Doudna groups (shown on the right). Figure 11 Structure of a domain of the HCV IRES RNA solved by NMR. The secondary structure of the 100 kDa IRES is shown, with domain IIb highlighted. The RNA adopts a loop E motif, with an S-turn in the backbone, as shown in the superposition of final NMR structures. HCV is an important health problem, and infects more the 2% of the worlds population. HCV is an positive-strand RNA virus, and its RNA genome is translated immediately after uncoating of the virus in the cytoplasm of infected (liver) cells. An internal ribosome entry site (IRES) mediates the initiation of translation of the HCV genome. The 40S ribosomal subunit binds directly to the IRES RNA structure, and thus avoids the need for a 5â cap and scanning; upon IRES binding, the start codon for translation is positioned near the P site. We have been studying the mechanism and structure of the HCV IRES. We have previously delineated the domains of the IRES that are critical for 40S subunit binding, and the proteins on the surface of the 40S subunit that mediate this interaction. Our current interests include: 1. Determination of the structure of the IRES using NMR spectroscopy. The IRES is a 100 kDa RNA, and we are using novel NMR approaches, including residual dipolar couplings. 2. High resolution NMR structures of isolated IRES RNA domains. 3. Determining the conformational changes that the IRES undergoes on 40S subunit binding. 4. Understanding the mechanism of IRES-mediated initiation. What are the order of bidning events How do initiation factors influence the process What is the rate of initiation How does IRES conformation affect initiation rate • RNA-protein interactions in HIV virus. Figure 12 The Tat-TAR protein RNA complex regulates transcription of HIV and BIV RNAs. The TAR element is located at the 5â end of both retroviral RNAs. In HIV, the Tat protein interacts with a trinucleotide bulge, and a host cyclin interacts with a hairpin loop. In BIV, the Tat protein binds with high affinity to the RNA site, and cyclin does not interact with the hairpin looo. (right) Three-dimensional structure of the HIV Tat-TAR interaction solved by NMR. Superposition of final NMR structures of the BIV Tat-peptide-TAR RNA complex solved using 13C, 15N-labeled BIV peptide. The peptide forms a beta turn within the RNA major groove. Figure 13 A host tRNALys3 primes reverse transcription of HIV genomic RNA. A binary complex of tRNA and genomic RNA is specifically recognized by HIV reverse transcriptase. We are studying the structure of the RNA-RNA complex using NMR. Figure 14 HIV genomic RNA and tRNA form a high-affinity complex. Imino proton NMR spectrum of free tRNALys3 and a 1:1 complex (50 kDa total) with HIV genomic RNA. RNA plays a criticial role in the HIV virus replication cycle. We have a longstanding interest in critical protein-RNA interactions that occur in HIV biology. First, the Tat protein is a transcriptional regulator coded by the HIV virus. It binds to a stem-loop structure at the 5â-end of HIV mRNAs, and binding to the nascent transcript stimulates transcription. We have studied the HIV and Bovine immunodeficiency virus (BIV) Tat-TAR interaction using peptides that correspond to the RNA-binding domains. We have refined the structure of the BIV Tat-TAR interaction using modern heteronuclear NMR methods. The protein adopts a beta-turn conformation upon RNA binding. We are using this as a template to design small molecule inhibitors of the Tat-TAR interaction. Current interests include: 1. Design of cyclic peptide mimics of the BIV Tat peptide that recognized TAR RNA. 2. Design of cyclic peptides that interact with HIV TAR RNA. A complex between a human tRNALys and HIV genomic RNA is the priming site for reverse transcription of the HIV RNA genome. HIV reverse transcriptase (RT) specifically recognizes this RNA-RNA complex. We are investigating the structural basis for initiation of RT using a combination of methods. The results of this work may have important ramifications for the mechanism of action of RT inhibitors. Our current work focuses on: 1. NMR structure determination of the 50 kDa tRNALys3-genomic RNA binary complex. 2. x-ray crystallographic analys of the RT initiation complex 3. The use of NMR to study the RT initiation complex. Joseph Puglisi Professor and Chair, Department of Structural Biology Director, Stanford Magnetic Resonance Laboratory ph fx Mailing Address: Stanford University School of Medicine D105 Fairchild Science Building 299 Campus Drive West Stanford, CA Manolia Margaris Administrative Associate ph fx GENTAMICINA. Complesso gentamicina-RNA target.")
38
Aminoglicosidi Gli aminoglicosidi agiscono legandosi irreversibilmente alla subunità ribosomiale 30 S e bloccando, di conseguenza, la sintesi proteica Questi antibiotici determinano anche un’alterata lettura dell’mRNA portando all’incorporazione di aminoacidi errati e causando, quindi, la produzione di proteine anomale o non funzionali. Per questa sua azione la streptomicina è in grado di sopprimere alcune mutazioni che di per sé sarebbero letali per il batterio, il quale può, pertanto, replicarsi solo in presenza di streptomicina (streptomicino-dipendenza). Sembra esservi una stretta correlazione tra attività battericida e capacità di indurre un’errata lettura dell’mRNA
. Sembra esservi una stretta correlazione tra attività battericida e capacità di indurre un’errata lettura dell’mRNA.")
39
Tetracicline Le tetracicline sono un gruppo di antibiotici prodotti da Streptomiceti e caratterizzati da una struttura molecolare tetraciclica Le tetracicline sono dotate di azione batteriostatica nei confronti di batteri sia Gram-positivi, sia Gram-negativi anaerobi e differenti microrganismi (la dizione “antibiotici ad ampio spettro” è stata usata per la prima volta nei loro riguardi). Le tetracicline sono un vasto gruppo di farmaci antibatterici inibitori della sintesi proteica. Efficaci contro i batteri Gram-positivi, Gram-negativi anaerobi e microrganismi come rickettsie, clamidie, micoplasmi, brucelle e diarree causate da Escherichia coli. Tetracycline antibiotics are protein synthesis inhibitors, inhibiting the binding of aminoacyl-tRNA to the mRNA-ribosome complex. They do so mainly by binding to the 30S ribosomal subunit in the mRNA translation complex. Tetracyclines also have been found to inhibit matrix metalloproteinases. This mechanism does not add to their antibiotic effects, but has led to extensive research on chemically modified tetracyclines or CMTs (like incyclinide) for the treatmet of rosacea, acne and various types of neoplasms.[6][7] Since incyclinide was announced to be ineffective for rosacea in September 2007, no drugs of this group will be marketed in the near future.[8]
. Le tetracicline sono un vasto gruppo di farmaci antibatterici inibitori della sintesi proteica. Efficaci contro i batteri Gram-positivi, Gram-negativi anaerobi e microrganismi come rickettsie, clamidie, micoplasmi, brucelle e diarree causate da Escherichia coli. Tetracycline antibiotics are protein synthesis inhibitors, inhibiting the binding of aminoacyl-tRNA to the mRNA-ribosome complex. They do so mainly by binding to the 30S ribosomal subunit in the mRNA translation complex. Tetracyclines also have been found to inhibit matrix metalloproteinases. This mechanism does not add to their antibiotic effects, but has led to extensive research on chemically modified tetracyclines or CMTs (like incyclinide) for the treatmet of rosacea, acne and various types of neoplasms.[6][7] Since incyclinide was announced to be ineffective for rosacea in September 2007, no drugs of this group will be marketed in the near future.[8]")
40
La loro azione antibatterica è dovuta ad un blocco della sintesi proteica in una fase molto iniziale: esse si legano alla subunità ribosomiale 30S subito dopo il legame dell’mRNA, impedendovi l’attacco dell’aminoacil-tRNA a livello del codone di inizio e bloccando la formazione del polisoma

41
Tetracicline OXYTETRACYCLINE TETRACICLINA ROLITETRACYCLINE
MECLOCYCLINE TETRACICLINA ROLITETRACYCLINE MECLOCYCLINE (INN) is a a tetracycline antibiotic. It is used topically (i.e. for skin infections) as it is totally insoluble in water and may cause liver and kidney damage if given systemically. Rolitetracycline is a tetracycline antibiotic. Tetracycline is N-Mannich base prodrug that is prepared from tetracycline by condensation with pyrrolidine and formaldehyde to produce rolitetracycline
 is a a tetracycline antibiotic. It is used topically (i.e. for skin infections) as it is totally insoluble in water and may cause liver and kidney damage if given systemically. Rolitetracycline is a tetracycline antibiotic. Tetracycline is N-Mannich base prodrug that is prepared from tetracycline by condensation with pyrrolidine and formaldehyde to produce rolitetracycline.")
42
6-demethyl-6-deoxytetracycline
Relazioni strutture-attività R1 - R4 sono le funzioni che possono essere modificate senza una perdita significativa d’attività A B C D Functional groups at positions 5, 6, and 7 may be removed without drastically altering the antimicrobial properties. The right configuration at C-5a and C-4 is essential for activity. Equilibration involving C-4 leads to the relatively inactive ) [25]. The hypothesis has been advanced that the principal active center is the C(11), C(12) diketone system of rings B and C [26,27,8]. 4-EPI-TETRACYCLINES (QUATRIMYCINS) 6-demethyl-6-deoxytetracycline (SANCYCLINE) La corretta configurazione nelle posizione C-5a e C-4 è esenciale per l’attività Epimerizzazione in C-4 da luogo a composti inattivi (quatrimicine) Frammento minimo per l’attività
 [25]. The hypothesis has been advanced that the principal active center is the C(11), C(12) diketone system of rings B and C [26,27,8]. 4-EPI-TETRACYCLINES (QUATRIMYCINS) 6-demethyl-6-deoxytetracycline. (SANCYCLINE) La corretta configurazione nelle posizione. C-5a e C-4 è esenciale per l’attività. Epimerizzazione in C-4 da luogo. a composti inattivi (quatrimicine) Frammento minimo per l’attività.")
43
Cloramfenicolo Il cloramfenicolo inibisce la sintesi proteica legandosi alla subunità 50S in corrispondenza del sito peptidiltransferasico inibendo la formazione del legame peptidico. Tuttavia sono sensibili all’azione del cloramfenicolo anche i ribosomi mitocondriali; ciò determina l’inibizione da parte dell’antibiotico di cellule eucariotiche in rapida crescita e con elevata attività mitocondriale, quali le cellule staminali del midollo osseo Il Cloramfenicolo (o Cloranfenicolo) è un antibiotico prodotto dal batterio Streptomyces venezuelae, scoperto nel 1947 ed attualmente ottenuto per sintesi chimica. Il cloranfenicolo presenta all' interno della sua struttura chimica due centri chirali, con la possibile formazione di 4 diversi stereoisomeri, in cui solo il D(-)treo, presenta un' efficacia antibatterica. Il cloranfenicolo ha efficacia contro un'estesa varietà di microrganismi, ma per via dei seri effetti collaterali (come i danni al midollo osseo, inclusa l'anemia aplastica), il suo impiego negli esseri umani viene preferibilmente limitato al trattamento d'infezioni gravi, con rischio di morte del paziente e per le quali non esista un'alternativa di pari efficacia (ad esempio per la febbre tifoide). L'OMS sostiene il suo uso nel trattamento di casi pediatrici nonostante i gravi effetti collaterali in diverse nazioni del Terzo mondo in assenza di alternative più economiche e dunque accessibili. Trova impiego per la cura del colera, poiché colpisce i vibrioni e riduce la diarrea, risultando efficace anche nei ceppi resistenti alla tetraciclina. Viene anche usato in gocce o in forma di unguento (in associazione con idrocortisone acetato) per il trattamento di congiuntiviti o dermatite accompagnate da sovrainfezioni batteriche. Viene spesso (illegalmente) usato in apicoltura per il controllo dell'agente della peste americana Paenibacillus larvae.
 è un antibiotico prodotto dal batterio Streptomyces venezuelae, scoperto nel 1947 ed attualmente ottenuto per sintesi chimica. Il cloranfenicolo presenta all interno della sua struttura chimica due centri chirali, con la possibile formazione di 4 diversi stereoisomeri, in cui solo il D(-)treo, presenta un efficacia antibatterica. Il cloranfenicolo ha efficacia contro un estesa varietà di microrganismi, ma per via dei seri effetti collaterali (come i danni al midollo osseo, inclusa l anemia aplastica), il suo impiego negli esseri umani viene preferibilmente limitato al trattamento d infezioni gravi, con rischio di morte del paziente e per le quali non esista un alternativa di pari efficacia (ad esempio per la febbre tifoide). L OMS sostiene il suo uso nel trattamento di casi pediatrici nonostante i gravi effetti collaterali in diverse nazioni del Terzo mondo in assenza di alternative più economiche e dunque accessibili. Trova impiego per la cura del colera, poiché colpisce i vibrioni e riduce la diarrea, risultando efficace anche nei ceppi resistenti alla tetraciclina. Viene anche usato in gocce o in forma di unguento (in associazione con idrocortisone acetato) per il trattamento di congiuntiviti o dermatite accompagnate da sovrainfezioni batteriche. Viene spesso (illegalmente) usato in apicoltura per il controllo dell agente della peste americana Paenibacillus larvae.")
44
Macrolidi e Lincosamidi
I macrolidi costituiscono una ampia famiglia di prodotti naturali (molti dei quali presentano attività antibiotica) costituiti da un anello lattonico macrociclico, tipicamente a 12, 14 o 16 termini. Il macrociclo può presentare numerose ramificazioni metiliche e lo stesso macrociclo può essere legato, mediante legame glicosidicoa due o più desossi zuccheri che raramente si ritrovano in altri composti. I più comuni zuccheri tipici dei macrolidi sono il L-cladinosio, L-micarosio, D-micinosio oppure L-oleandrosio. Almeno una unità saccaridica deve essere un amminozucchero come D-desossamina o D-forosamina Il loro spettro d’azione è poco più ampio di quello delle penicilline (includendo anche alcuni Gram-negativi) ed hanno azione batteriostatica. I lincosamidi sono antibiotici batteriostatici con un meccanismo e spettro d’azione simile a quello dei macrolidi, pur avendo una struttura chimica molto diversa. macrolidi hanno uno spettro d' azione simile a quello delle penicilline. Di norma si preferisce l' uso dei β-lattamici ma si possono usare anche i macrolidi come validi sostituti soprattutto nel caso in cui il paziente sia allergico o insensibile alle penicilline
 costituiti da un anello lattonico macrociclico, tipicamente a 12, 14 o 16 termini. Il macrociclo può presentare numerose ramificazioni metiliche e lo stesso macrociclo può essere legato, mediante legame glicosidicoa due o più desossi zuccheri che raramente si ritrovano in altri composti. I più comuni zuccheri tipici dei macrolidi sono il L-cladinosio, L-micarosio, D-micinosio oppure L-oleandrosio. Almeno una unità saccaridica deve essere un amminozucchero come D-desossamina o D-forosamina Il loro spettro d’azione è poco più ampio di quello delle penicilline (includendo anche alcuni Gram-negativi) ed hanno azione batteriostatica. I lincosamidi sono antibiotici batteriostatici con un meccanismo e spettro d’azione simile a quello dei macrolidi, pur avendo una struttura chimica molto diversa. macrolidi hanno uno spettro d azione simile a quello delle penicilline. Di norma si preferisce l uso dei β-lattamici ma si possono usare anche i macrolidi come validi sostituti soprattutto nel caso in cui il paziente sia allergico o insensibile alle penicilline.")
45
Macrolidi e Lincosamidi
CLARITROMICINA ERITROMICINA L'eritromicina è il capostipite di un gruppo di antibiotici denominati macrolidi. Entrata in uso nel 1953, il suo uso è ancora molto diffuso. I batteri sensibili a questa classe di antibiotici e pertanto compresi nello spettro d'azione dell'eritromicina sono i cocchi Gram-positivi, come lo Staphylococcus aureus, pneumococchi, streptococchi e cocchi Gram-negativi. È inoltre attiva su microrganismi quali Bordetella pertussis, Brucella, Mycoplasma pneumoniae ed anche organismi intracellulari come la Rickettsia. Per quanto riguarda la struttura chimica dell'eritromicina, la funzione chetonica risulta molto importante per la sua attività. La claritromicina è un antibatterico generale appartenente al gruppo dei macrolidi, viene impiegato nel trattamento di diversi disturbi. Ha emivita di circa 6 ore, è acidostabile dunque non necessita di essere incapsulata (come la eritromicina) per essere somministrata in forma orale, nelle urine viene eliminata per il 60%. The first lincosamide to be discovered was lincomycin isolated from Streptomyces lincolnensis. Lincomycin has been superseded by clindamycin, which exhibits improved antibacterial activity. Clindamycin also exhibits some activity against parasitic protozoa, and has been used in toxoplasmosis and malaria. They are normally used to treat staphylococci and streptococci, and have proved useful in treating Bacteroides fragilis and some other anaerobes. They are used in the treatment of Toxic Shock Syndrome and thought to directly block the M protein production that leads to the severe inflammatory response. CLINDAMYCIN LINCOMYCIN
 per essere somministrata in forma orale, nelle urine viene eliminata per il 60%. The first lincosamide to be discovered was lincomycin isolated from Streptomyces lincolnensis. Lincomycin has been superseded by clindamycin, which exhibits improved antibacterial activity. Clindamycin also exhibits some activity against parasitic protozoa, and has been used in toxoplasmosis and malaria. They are normally used to treat staphylococci and streptococci, and have proved useful in treating Bacteroides fragilis and some other anaerobes. They are used in the treatment of Toxic Shock Syndrome and thought to directly block the M protein production that leads to the severe inflammatory response. CLINDAMYCIN. LINCOMYCIN.")
46
I macrolidi inibiscono la sintesi proteica agendo di conseguenza come batteriostatico. Nello specifico si legano alla subunità 50s del ribosoma batterico bloccando il meccanismo di traslocazione dei t-RNA che portano gli amminoacidi all'interno del ribosoma, impedendo che passino dal sito di ingresso A, al sito P e quindi al sito di uscita E. L'eritromicina,impedendo così la sintesi delle proteine.

47
Acido fusidico L’acido fusidico è un antibioico steroideo che esplica la sua azione di tipo batteriostatico interagendo con il fattore extraribosomiale G (EF-G) necessario per il processo di allungamento della catena polipeptidica In presenza di acido fusidico si ha la stabilizzazione del complesso EF-G/GTP/ribosoma, con conseguente impedimento della traslocazione in quanto il fattore di allungamento non è utilizzabile perché legato all’antibiotico e il ribosoma è incapace di muoversi lungo l’mRNA The steroid antibiotic fusidic acid is used to treat Gram-positive infections. It acts by preventing translocation of peptidyl tRNA. Resistant mutants may easily be selected, even during therapy and therefore fusidic acid is usually administered in combination with another antibiotic. This helps reduce the risk of selecting resistant mutants. To survive, the fusidic acid resistant mutants must also become resistant to the antibiotic given in combination. If the chance of selecting fusidic acid resistance is 10-5 and that of chromosomal mutation to resistance to the second agent is 10-8 then the theoretical chance of a double mutant arising from combination therapy is (=10-5 x 10-8). This is so low as to be considered insignificant but, in practice, the chance of acquiring resistance to the second agent is increased by the presence of mobile bacterial genes encoding resistance. These can be acquired more easily than chromosomal genes can mutate to confer a resistance phenotype on a bacterium.
 necessario per il processo di allungamento della catena polipeptidica. In presenza di acido fusidico si ha la stabilizzazione del complesso EF-G/GTP/ribosoma, con conseguente impedimento della traslocazione in quanto il fattore di allungamento non è utilizzabile perché legato all’antibiotico e il ribosoma è incapace di muoversi lungo l’mRNA. The steroid antibiotic fusidic acid is used to treat Gram-positive infections. It acts by preventing translocation of peptidyl tRNA. Resistant mutants may easily be selected, even during therapy and therefore fusidic acid is usually administered in combination with another antibiotic. This helps reduce the risk of selecting resistant mutants. To survive, the fusidic acid resistant mutants must also become resistant to the antibiotic given in combination. If the chance of selecting fusidic acid resistance is 10-5 and that of chromosomal mutation to resistance to the second agent is 10-8 then the theoretical chance of a double mutant arising from combination therapy is (=10-5 x 10-8). This is so low as to be considered insignificant but, in practice, the chance of acquiring resistance to the second agent is increased by the presence of mobile bacterial genes encoding resistance. These can be acquired more easily than chromosomal genes can mutate to confer a resistance phenotype on a bacterium.")
48
Ossazolidinone Ossazolidinoni ripresentano una nuova classe d’antibiotici Linezolid, capostipite degli oxazolidinoni, è il primo antibiotico innovativo da alcuni decenni a questa parte. Attua inibendo la formazione di un complesso di inizio funzionale 70S nella subunità ribosomiale batterica 50S Linezolid è attivo nei confronti dei cocchi Gram+ con resistenza multipla, tra cui MRSA ( Staphylococcus aureus Meticillina-resistente ), VRSA ( Staphylococcus aureus Vancomicina-resistente ), enterococchi Vancomicina-resistenti; il suo spettro d’attività comprende anche alcuni batteri anaerobi. Linezolid Oxazolidinone antibiotics represent the first new class of antibiotics for clinical usage within the past 30 yearsLinezolid è un principio attivo per uso ospedaliero, è attivo su batteri Gram+ aerobi e anaerobi. Attivo anche su MRSA (ceppi di Stafilococco [[meticillina|meticillino-resistestenti) e VRSA (ceppi di Stafilococco vancomicina-resistenti). Due formulazioni: sacche, pronte per l'uso, da 300 ml contenenti 600 mg di principio attivo e compresse da 600 mg. Le due formulazioni sono bioequivalenti (stessa efficacia). Indicazioni terapeutiche: infezioni sostenute da Staphylococcus, Enterococcus, Listeria monocytogenes e Clostridium difficile resistenti ad altri trattamenti, polmoniti nosocomiali e polmoniti contratte in comunità. Diffonde rapidamente(poche ore dalla fine della prima somministrazione) in tutti gli organi e tessuti grazie al suo volume di distribuzione di circa 50 litri (che corrisponde alla nostra acqua corporea totale). Il suo indice di penetrazione tissutale è più alto di altri antibiotici. La famiglia a cui appartiene è quella degli oxazolidinoni di cui è, attualmente, l'unico rappresentante. Da considerare come il superamento, in termini di efficacia, rapidità d'azione e sicurezza, dei glicopeptidi (teicoplanina e vancomicina). Linezolid non necessita di aggiustamento posologico (sempre 1 somministrazione ogni 12 ore) anche nei pazienti con insufficienza epatica e/o renale
, VRSA ( Staphylococcus aureus Vancomicina-resistente ), enterococchi Vancomicina-resistenti; il suo spettro d’attività comprende anche alcuni batteri anaerobi. Linezolid. Oxazolidinone antibiotics represent the first new class of antibiotics for clinical usage within the past 30 yearsLinezolid è un principio attivo per uso ospedaliero, è attivo su batteri Gram+ aerobi e anaerobi. Attivo anche su MRSA (ceppi di Stafilococco [[meticillina|meticillino-resistestenti) e VRSA (ceppi di Stafilococco vancomicina-resistenti). Due formulazioni: sacche, pronte per l uso, da 300 ml contenenti 600 mg di principio attivo e compresse da 600 mg. Le due formulazioni sono bioequivalenti (stessa efficacia). Indicazioni terapeutiche: infezioni sostenute da Staphylococcus, Enterococcus, Listeria monocytogenes e Clostridium difficile resistenti ad altri trattamenti, polmoniti nosocomiali e polmoniti contratte in comunità. Diffonde rapidamente(poche ore dalla fine della prima somministrazione) in tutti gli organi e tessuti grazie al suo volume di distribuzione di circa 50 litri (che corrisponde alla nostra acqua corporea totale). Il suo indice di penetrazione tissutale è più alto di altri antibiotici. La famiglia a cui appartiene è quella degli oxazolidinoni di cui è, attualmente, l unico rappresentante. Da considerare come il superamento, in termini di efficacia, rapidità d azione e sicurezza, dei glicopeptidi (teicoplanina e vancomicina). Linezolid non necessita di aggiustamento posologico (sempre 1 somministrazione ogni 12 ore) anche nei pazienti con insufficienza epatica e/o renale.")
49
Antibiotici attivi sulla replicazione del DNA:
Inibitori della DNA-girase batterica

50
La girasi batterica è formata da due coppie di identici polipeptidi, denominati subunità A e B. Le subunità A tagliano la catena di DNA in siti specifici e ne consentono lo srotolamento, mentre le subunità B sono responsabili della spiralizzazione negativa del DNA. Ambedue le funzioni sono necessarie per ottenere le modificazioni topologiche indispensabili alla produzione della “forca replicativa” e, quindi, alla replicazione del DNA Gyrase is present in prokaryotes and some eukaryotes, but the enzymes are not entirely similar in structure or sequence, and have different affinities for different molecules. It is not present in humans. This makes gyrase a good target for antibiotics. Two classes of antibiotics that inhibit gyrase are: The aminocoumarins (including novobiocin). Aminocoumarins work by competitive inhibition of energy transduction of DNA gyrase by binding to the ATPase active site located on the GyrB subunit. The quinolones (including nalidixic acid and ciprofloxacin). Quinolones bind these enzymes and prevent them from decatenating replicating DNA. Quinolone-resistant bacteria frequently harbor mutated topoisomerases that resist quinolone binding.
. Aminocoumarins work by competitive inhibition of energy transduction of DNA gyrase by binding to the ATPase active site located on the GyrB subunit. The quinolones (including nalidixic acid and ciprofloxacin). Quinolones bind these enzymes and prevent them from decatenating replicating DNA. Quinolone-resistant bacteria frequently harbor mutated topoisomerases that resist quinolone binding.")
52
I chinoloni I chinoloni sono una classe di composti antibatterici sintetici che inattivano la girasi interferendo con la subunità A …….. e la novobiocina L’antibiotico novobiocina ottiene lo stesso risultato interferendo con la subunità B. L’azione antibatterica dei chinoloni è sinergica con quella della novobiocina (l’inattivazione della girasi è potenziata se sono contemporaneamente colpite le due subunità costitutive) e non presenta resistenza crociata. sf. [da novo+bio-+(-mi)cina]. Antibiotico isolato da colture di Streptomyces spheroides. Ha uno spettro d'azione molto simile a quello della penicillina, agendo soprattutto sugli stafilococchi e sui pneumococchi con effetti battericidi o batteriostatici secondo le dosi. La novobiocina viene impiegata nel trattamento di infezioni da germi resistenti alla penicillina, oppure nei soggetti allergici alla penicillina.
 e non presenta resistenza. crociata. sf. [da novo+bio-+(-mi)cina]. Antibiotico isolato da colture di Streptomyces spheroides. Ha uno spettro d azione molto simile a quello della penicillina, agendo soprattutto sugli stafilococchi e sui pneumococchi con effetti battericidi o batteriostatici secondo le dosi. La novobiocina viene impiegata nel trattamento di infezioni da germi resistenti alla penicillina, oppure nei soggetti allergici alla penicillina.")
53
I chinoloni La storia degli antibatterici chinolonici ha avuto origine con una scoperta casuale all’inizio degli anni sessanta. Nel corso di una sintesi alternativa della clorochina, i chimici della Sterling- Winthrop isolarono come sottoprodotto il composto 1 (Figura 1), che risultò provvisto di una certa attività antibatterica. Ripetute modificazioni chimiche di questo lead permisero l’ottenimento di una varietà di analoghi strutturali che furono saggiati come antibatterici, arrivando così alla scoperta dell’acido nalidissico (Figura 1). Questo composto, dotato di moderata attività contro i germi gramnegativi, ha rappresentato il capostipite della famiglia dei chinoloni e fu introdotto in terapia nel 1963 Un progresso sensibile nel settore dei chinoloni si ebbe nel 1980, allorché i chimici della Kyorin misero a punto la preparazione della norflossacina, derivante dalla combinazione di elementi 2strutturali dell’acido pipemidico (7-piperazina) e della flumechina (6-fluoro) (Figura 3). La norflossacina mostra una certa attività sui gram-positivi oltre ad una attività contro i gram-negativi superiore rispetto agli agenti precedenti, ma in pratica è ancora indicata soltanto per le infezioni del tratto urinario ed il trattamento di prostatiti ed infezioni trasmesse sessualmente (gonorrea), a causa dei suoi bassi livelli ematici e della scarsa distribuzione tissutale. La norflossacina è stato il primo fluorochinolone propriamente detto in quanto presenta non solo il fluoro in posizione 6, ma anche l’altro elemento strutturale fondamentale che è la piperazina in 7 (si dovrebbe forse parlare di fluoropiperazochinoloni). come chemioterapico delle vie urina
, che risultò provvisto di una certa. attività antibatterica. Ripetute modificazioni chimiche di questo lead permisero l’ottenimento di una. varietà di analoghi strutturali che furono saggiati come antibatterici, arrivando così alla scoperta. dell’acido nalidissico (Figura 1). Questo composto, dotato di moderata attività contro i germi gramnegativi, ha rappresentato il capostipite della famiglia dei chinoloni e fu introdotto in terapia nel Un progresso sensibile nel settore dei chinoloni si ebbe nel 1980, allorché i chimici della. Kyorin misero a punto la preparazione della norflossacina, derivante dalla combinazione di elementi. 2strutturali dell’acido pipemidico (7-piperazina) e della flumechina (6-fluoro) (Figura 3). La. norflossacina mostra una certa attività sui gram-positivi oltre ad una attività contro i gram-negativi. superiore rispetto agli agenti precedenti, ma in pratica è ancora indicata soltanto per le infezioni del. tratto urinario ed il trattamento di prostatiti ed infezioni trasmesse sessualmente (gonorrea), a causa dei. suoi bassi livelli ematici e della scarsa distribuzione tissutale. La norflossacina è stato il primo. fluorochinolone propriamente detto in quanto presenta non solo il fluoro in posizione 6, ma anche. l’altro elemento strutturale fondamentale che è la piperazina in 7 (si dovrebbe forse parlare di. fluoropiperazochinoloni). come chemioterapico delle vie urina.")
54
Chinoloni terza e quarta generazione
TEMAFLOXACINA Questi nuovi composti (Figura 4) sono caratterizzati da aumentata novità e complessità strutturale, che hanno determinato nuove ed utili proprietà biologiche, quali attività contro cocchi gram-positivi (ed in particolare S. pneumoniae) e, in certi composti della serie, contro anaerobi e patogeni atipici. In alcuni casi la maggior potenza di azione e l’ampiezza dello spettro di azione si coniuga con proprietà farmacocinetiche più favorevoli che permettono di somministrare questi composti una sola volta al dì. In Figura 5 sono riportate schematicamente le variazioni strutturali principali dei chinoloni di terza generazione e dei composti attualmente in sviluppo. In sintesi, la sostituzione in 8 (alogeno o metossi) incrementa generalmente l’attività sui gram-positivi e negativi, probabilmente migliorando la capacità di penetrazione della celllula batterica. Tuttavia, l’aumento di lipofilia che è stato attuato nei chinoloni di terza generazione tende ad aumentare di solito più l’azione sui gram-positivi che quella sui gram-negativi. Ciò può essere conseguenza degli effetti opposti che la idrofobicità ha sulla penetrazione: l’aumento del logP tende ad aumentare l’accumulo nello S. aureus diminuendo, invece, la penetrazione nei gram-negativi. La lipofilia può essere aumentata anche con l’introduzione di sostituenti alchilici al C5 e/o nel nucleo piperazinico, come pure con la sostituzione della piperazina con 3-amminopirrolidine. E’ bene puntualizzare che, sebbene certe sostituzioni possano migliorare per se una particolare proprietà biologica o chimica, le caratteristiche complessive di una molecola derivano tuttavia dalla interazione di tutti i sostituenti uno con l’altro e con lo specifico nucleo utilizzato
 sono caratterizzati da aumentata novità e complessità. strutturale, che hanno determinato nuove ed utili proprietà biologiche, quali attività contro cocchi. gram-positivi (ed in particolare S. pneumoniae) e, in certi composti della serie, contro anaerobi e. patogeni atipici. In alcuni casi la maggior potenza di azione e l’ampiezza dello spettro di azione si. coniuga con proprietà farmacocinetiche più favorevoli che permettono di somministrare questi. composti una sola volta al dì. In Figura 5 sono riportate schematicamente le variazioni strutturali principali dei chinoloni di. terza generazione e dei composti attualmente in sviluppo. In sintesi, la sostituzione in 8 (alogeno o. metossi) incrementa generalmente l’attività sui gram-positivi e negativi, probabilmente migliorando la. capacità di penetrazione della celllula batterica. Tuttavia, l’aumento di lipofilia che è stato attuato nei. chinoloni di terza generazione tende ad aumentare di solito più l’azione sui gram-positivi che quella sui. gram-negativi. Ciò può essere conseguenza degli effetti opposti che la idrofobicità ha sulla. penetrazione: l’aumento del logP tende ad aumentare l’accumulo nello S. aureus diminuendo, invece, la penetrazione nei gram-negativi. La lipofilia può essere aumentata anche con l’introduzione di. sostituenti alchilici al C5 e/o nel nucleo piperazinico, come pure con la sostituzione della piperazina. con 3-amminopirrolidine. E’ bene puntualizzare che, sebbene certe sostituzioni possano migliorare per se una particolare. proprietà biologica o chimica, le caratteristiche complessive di una molecola derivano tuttavia dalla. interazione di tutti i sostituenti uno con l’altro e con lo specifico nucleo utilizzato.")
55
Analisi strutturale dei antibiotici quinolonici

56
FARMACOFORO Sostituenti ottimali alle varie posizioni.
SITO DI LEGAME AL DNA DOMINIO DI AUTOASSEMBLAGGIO SITO DI LEGAME ALL’ENZIMA 1 2 3 1 4 5 6 7 1 8 FARMACOFORO Rifampicin (INN) (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from Rifampicin (INN) (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from Amycolatopsis rifamycinica (formerly known as Amycolatopsis mediterranei and Streptomyces mediterranei).[2] Rifampicin may be abbreviated R, RMP, RD, RA, or RIF (US). In 1957, a sample of soil coming from a pine wood on the French Riviera was brought for analysis to the Lepetit Pharmaceuticals research lab in Milan, Italy. There, a research group headed by Prof. Piero Sensi (1920-) discovered a new bacterium. This new species appeared immediately of great scientific interest since it was producing a new class of molecules with antibiotic activity. Because Prof. Sensi and some of his fellow researchers were particularly fond of the French crime story Rififi (about a jewel heist and rival gangs),[3] they decided to call these compounds "Rifamycins". After two years of attempts in order to obtain more stable semi-synthetic products, in 1959 a new molecule with high efficacy and good tolerability was produced and was named "Rifampicin". It is also known as rifaldazine, R/AMP, and Rofact (in Canada). There are various types of rifamycins from which this is derived, but this particular form, with a 4-methyl-1-piperazinaminyl group, is by far the most clinically effective. Rifampicin is an intensely red solid, and the small fraction which reaches body fluids is known for imparting a harmless red-orange color to the urine (and to a lesser extent, also sweat and tears) of users, for a few hours after a dose. Maximal concentrations in the blood are decreased by about a third when the antibiotic is taken with food. [4]. Rifampicin was introduced in 1967,[5] as a major addition to the cocktail-drug treatment of tuberculosis and inactive meningitis, along with isoniazid, ethambutol, pyrazinamide and streptomycinRifampicin is typically used to treat Mycobacterium infections, including tuberculosis and leprosy. With multidrug therapy used as the standard treatment of leprosy, rifampicin is always used in combination with dapsone and clofazimine to avoid eliciting drug resistance. Posizione 1: questa posizione è coinvolta nell’assemblaggio di due o più molecole di farmaco. Un sostituente ciclopropilico è considerato in questa posizione la modificazione che esalta di più la potenza, seguita dal sostituente 2,4-difluorofenile. Altri sostituenti possono abbassare la capacità della molecola di legarsi alla tasca dell’enzima e quindi la potenza. L’isomero S della ofloxacina (levofloxacina), anche se presenta una potenza minore della molecola col gruppo ciclopropilico, grazie all’anello che connette le posizioni 1 e 8 e crea una struttura triciclica, è un’utile alternativa. Posizione 2: questa posizione è molto vicino al sito di legame per la DNA girasi o la topoisomerasi IV, così si pensa che ogni gruppo voluminoso inibisce l’accesso al sito di legame e diminuisce l’attività. Solo uno zolfo incorporato in un piccolo anello può rimpiazzare l’idrogeno nella posizione R2. Posizioni 3 e 4: queste due posizioni sul nucleo chinolonico sono considerate critiche per il legame al DNA e ancora non ci sono sostituzioni utili per questa posizione. Essenziali per l’attività antimicrobica sono i gruppi 3-carbossilico e 4-carbonilico. Posizione 5: i sostituenti in questa posizione sembrano avere la capacità di alterare la configurazione sterica planare della molecola. Aggiunta di gruppi di piccola grandezza come gruppi metilici, idrossilici, o amminici possono aumentare in modo marcato l’attività in vitro. Alogenuri e gruppi metossilici tendono a diminuire l’attività. Posizione 6: l’addizione di un atomo di fluoro ha molto migliorato l’attività antimicrobica dei primi agenti antibatterici chinolonici. Nuovi chinoloni idrogenati in posizione 6 stanno dando promettenti sviluppi. Mettendo in posizione 6 un gruppo amminico, la potenza delle molecole ottenute dipende dai sostituenti nelle posizioni 7 e 8. Posizione 7: questa posizione è considerata quella che direttamente agisce con la DNA girasi o con la topoisomerasi IV. I sostituenti ottimali in questa posizione sono gruppi che contengono, come minimo, eterocicli di 5 o 6 membri contenenti azoto, come gruppi ammonopirrolidinici (che aumentano l’attività verso batteri gram-positivi) e piperazinici (che aumentano l’attività verso batteri gram-negativi). Si è osservato recentemente che un volume maggiore nella posizione R7 (Moxifloxacina) aumenta l’attività nei confronti degli anaerobi e conferisce protezione dalle proteine del batterio che mandano fuori il farmaco, diminuendo la resistenza batterica. Posizione 8: questa posizione influenza, come la posizione 5, la configurazione sterica della molecola chinolonica e i cambi apportati alterano l’accesso del chinolone alla tasca dell’enzima o ai siti di legame sul DNA. E’ stato osservato che specifici cambi in questa posizione alterano il bersaglio dei fluorochinoloni. Infatti un idrogeno, come nella ciprofloxacina, o anche un anello fuso, come nell’ofloxacina e nella levofloxacina, porta ad una elevata attività nei confronti dell’enzima topoisomerasi IV, e ad un’attività clinicamente poco utile nei confronti della DNA girasi. Un alogeno non sostituito come F o Cl, migliora l’attività nei confronti degli organismi anaerobi e sposta l’attività nei confronti della DNA girasi, riducendo l’azione anti-topoisomerasica. L’aggiunta di gruppi metossilici e la sostituzione del C con un N migliora l’attività ed esalta la potenza sia contro la girasi che contro la topoisomerasi. Sostituenti ottimali alle varie posizioni.
 (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from. Rifampicin (INN) (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from Amycolatopsis rifamycinica (formerly known as Amycolatopsis mediterranei and Streptomyces mediterranei).[2] Rifampicin may be abbreviated R, RMP, RD, RA, or RIF (US). In 1957, a sample of soil coming from a pine wood on the French Riviera was brought for analysis to the Lepetit Pharmaceuticals research lab in Milan, Italy. There, a research group headed by Prof. Piero Sensi (1920-) discovered a new bacterium. This new species appeared immediately of great scientific interest since it was producing a new class of molecules with antibiotic activity. Because Prof. Sensi and some of his fellow researchers were particularly fond of the French crime story Rififi (about a jewel heist and rival gangs),[3] they decided to call these compounds Rifamycins . After two years of attempts in order to obtain more stable semi-synthetic products, in 1959 a new molecule with high efficacy and good tolerability was produced and was named Rifampicin . It is also known as rifaldazine, R/AMP, and Rofact (in Canada). There are various types of rifamycins from which this is derived, but this particular form, with a 4-methyl-1-piperazinaminyl group, is by far the most clinically effective. Rifampicin is an intensely red solid, and the small fraction which reaches body fluids is known for imparting a harmless red-orange color to the urine (and to a lesser extent, also sweat and tears) of users, for a few hours after a dose. Maximal concentrations in the blood are decreased by about a third when the antibiotic is taken with food. [4]. Rifampicin was introduced in 1967,[5] as a major addition to the cocktail-drug treatment of tuberculosis and inactive meningitis, along with isoniazid, ethambutol, pyrazinamide and streptomycinRifampicin is typically used to treat Mycobacterium infections, including tuberculosis and leprosy. With multidrug therapy used as the standard treatment of leprosy, rifampicin is always used in combination with dapsone and clofazimine to avoid eliciting drug resistance. Posizione 1: questa posizione è coinvolta nell’assemblaggio di due o più molecole di farmaco. Un sostituente ciclopropilico è considerato in questa posizione la modificazione che esalta di più la potenza, seguita dal sostituente 2,4-difluorofenile. Altri sostituenti possono abbassare la capacità della molecola di legarsi alla tasca dell’enzima e quindi la potenza. L’isomero S della ofloxacina (levofloxacina), anche se presenta una potenza minore della molecola col gruppo ciclopropilico, grazie all’anello che connette le posizioni 1 e 8 e crea una struttura triciclica, è un’utile alternativa. Posizione 2: questa posizione è molto vicino al sito di legame per la DNA girasi o la topoisomerasi IV, così si pensa che ogni gruppo voluminoso inibisce l’accesso al sito di legame e diminuisce l’attività. Solo uno zolfo incorporato in un piccolo anello può rimpiazzare l’idrogeno nella posizione R2. Posizioni 3 e 4: queste due posizioni sul nucleo chinolonico sono considerate critiche per il legame al DNA e ancora non ci sono sostituzioni utili per questa posizione. Essenziali per l’attività antimicrobica sono i gruppi 3-carbossilico e 4-carbonilico. Posizione 5: i sostituenti in questa posizione sembrano avere la capacità di alterare la configurazione sterica planare della molecola. Aggiunta di gruppi di piccola grandezza come gruppi metilici, idrossilici, o amminici possono aumentare in modo marcato l’attività in vitro. Alogenuri e gruppi metossilici tendono a diminuire l’attività. Posizione 6: l’addizione di un atomo di fluoro ha molto migliorato l’attività antimicrobica dei primi agenti antibatterici chinolonici. Nuovi chinoloni idrogenati in posizione 6 stanno dando promettenti sviluppi. Mettendo in posizione 6 un gruppo amminico, la potenza delle molecole ottenute dipende dai sostituenti nelle posizioni 7 e 8. Posizione 7: questa posizione è considerata quella che direttamente agisce con la DNA girasi o con la topoisomerasi IV. I sostituenti ottimali in questa posizione sono gruppi che contengono, come minimo, eterocicli di 5 o 6 membri contenenti azoto, come gruppi ammonopirrolidinici (che aumentano l’attività verso batteri gram-positivi) e piperazinici (che aumentano l’attività verso batteri gram-negativi). Si è osservato recentemente che un volume maggiore nella posizione R7 (Moxifloxacina) aumenta l’attività nei confronti degli anaerobi e conferisce protezione dalle proteine del batterio che mandano fuori il farmaco, diminuendo la resistenza batterica. Posizione 8: questa posizione influenza, come la posizione 5, la configurazione sterica della molecola chinolonica e i cambi apportati alterano l’accesso del chinolone alla tasca dell’enzima o ai siti di legame sul DNA. E’ stato osservato che specifici cambi in questa posizione alterano il bersaglio dei fluorochinoloni. Infatti un idrogeno, come nella ciprofloxacina, o anche un anello fuso, come nell’ofloxacina e nella levofloxacina, porta ad una elevata attività nei confronti dell’enzima topoisomerasi IV, e ad un’attività clinicamente poco utile nei confronti della DNA girasi. Un alogeno non sostituito come F o Cl, migliora l’attività nei confronti degli organismi anaerobi e sposta l’attività nei confronti della DNA girasi, riducendo l’azione anti-topoisomerasica. L’aggiunta di gruppi metossilici e la sostituzione del C con un N migliora l’attività ed esalta la potenza sia contro la girasi che contro la topoisomerasi. Sostituenti ottimali alle varie posizioni.")
57
Inibizione della sintesi di RNA: le rifampicine
Le rifampicine sono un gruppo di antibiotici isolati in Italia, dalle colture di Nocardia mediterranea, che agiscono legandosi direttamente alla subunità β dell’RNA polimerasi batterica che rendono non funzionale. Le rifampicine sono attive nei confronti dei batteri Gram-positivi e Gram-negativi, e in particolare nei confronti del micobatterio tubercolare per le cui infezioni rappresentano uno dei farmaci di elezione. Rifampicin (INN) (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from Rifampicin (INN) (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from Amycolatopsis rifamycinica (formerly known as Amycolatopsis mediterranei and Streptomyces mediterranei).[2] Rifampicin may be abbreviated R, RMP, RD, RA, or RIF (US). In 1957, a sample of soil coming from a pine wood on the French Riviera was brought for analysis to the Lepetit Pharmaceuticals research lab in Milan, Italy. There, a research group headed by Prof. Piero Sensi (1920-) discovered a new bacterium. This new species appeared immediately of great scientific interest since it was producing a new class of molecules with antibiotic activity. Because Prof. Sensi and some of his fellow researchers were particularly fond of the French crime story Rififi (about a jewel heist and rival gangs),[3] they decided to call these compounds "Rifamycins". After two years of attempts in order to obtain more stable semi-synthetic products, in 1959 a new molecule with high efficacy and good tolerability was produced and was named "Rifampicin". It is also known as rifaldazine, R/AMP, and Rofact (in Canada). There are various types of rifamycins from which this is derived, but this particular form, with a 4-methyl-1-piperazinaminyl group, is by far the most clinically effective. Rifampicin is an intensely red solid, and the small fraction which reaches body fluids is known for imparting a harmless red-orange color to the urine (and to a lesser extent, also sweat and tears) of users, for a few hours after a dose. Maximal concentrations in the blood are decreased by about a third when the antibiotic is taken with food. [4]. Rifampicin was introduced in 1967,[5] as a major addition to the cocktail-drug treatment of tuberculosis and inactive meningitis, along with isoniazid, ethambutol, pyrazinamide and streptomycinRifampicin is typically used to treat Mycobacterium infections, including tuberculosis and leprosy. With multidrug therapy used as the standard treatment of leprosy, rifampicin is always used in combination with dapsone and clofazimine to avoid eliciting drug resistance.
 (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from. Rifampicin (INN) (pronounced /rɪˈfæmpəsɪn/) or rifampin (USAN) is a bactericidal antibiotic drug of the rifamycin group.[1] It is a semisynthetic compound derived from Amycolatopsis rifamycinica (formerly known as Amycolatopsis mediterranei and Streptomyces mediterranei).[2] Rifampicin may be abbreviated R, RMP, RD, RA, or RIF (US). In 1957, a sample of soil coming from a pine wood on the French Riviera was brought for analysis to the Lepetit Pharmaceuticals research lab in Milan, Italy. There, a research group headed by Prof. Piero Sensi (1920-) discovered a new bacterium. This new species appeared immediately of great scientific interest since it was producing a new class of molecules with antibiotic activity. Because Prof. Sensi and some of his fellow researchers were particularly fond of the French crime story Rififi (about a jewel heist and rival gangs),[3] they decided to call these compounds Rifamycins . After two years of attempts in order to obtain more stable semi-synthetic products, in 1959 a new molecule with high efficacy and good tolerability was produced and was named Rifampicin . It is also known as rifaldazine, R/AMP, and Rofact (in Canada). There are various types of rifamycins from which this is derived, but this particular form, with a 4-methyl-1-piperazinaminyl group, is by far the most clinically effective. Rifampicin is an intensely red solid, and the small fraction which reaches body fluids is known for imparting a harmless red-orange color to the urine (and to a lesser extent, also sweat and tears) of users, for a few hours after a dose. Maximal concentrations in the blood are decreased by about a third when the antibiotic is taken with food. [4]. Rifampicin was introduced in 1967,[5] as a major addition to the cocktail-drug treatment of tuberculosis and inactive meningitis, along with isoniazid, ethambutol, pyrazinamide and streptomycinRifampicin is typically used to treat Mycobacterium infections, including tuberculosis and leprosy. With multidrug therapy used as the standard treatment of leprosy, rifampicin is always used in combination with dapsone and clofazimine to avoid eliciting drug resistance.")
58
Inibisce specificamente l’iniziazione della sintesi di RNA
Inibisce specificamente l’iniziazione della sintesi di RNA. Non blocca il binding RNA polymerase -DNA template ma interferisce con la formazione dei primi legami fosfodiester nella catena del RNA RIFAMPICINA La struttura del complesso RNA polimerase-rifampicina rivela che l’antibiotico blocca il canale nel quale sono allocati gli ibridi RNA-DNA generati per l’azione dell’enzime, bloccando, così l’elongazione della catena

59
CHEMIOTERAPICI ANTIMETABOLITA
Composto con struttura chimica simile a un metabolita (vitamina, ormone, amminoacido, cofattore, coenzima ecc.), con capacità antagonista rispetto alle sue funzioni, che induce segni di deficienza del metabolita stesso. Generalmente l’azione dell’antimetabolita si esplica con modalità competitiva rispetto al metabolita. L’azione antagonista di molti antimetabolita si ripercuote in vario modo sulla biosintesi e sulla funzione degli acidi nucleici e aminoacidi
, con capacità antagonista rispetto alle sue funzioni, che induce segni di deficienza del metabolita stesso. Generalmente l’azione dell’antimetabolita si esplica con modalità competitiva rispetto al metabolita. L’azione antagonista di molti antimetabolita si ripercuote in vario modo sulla biosintesi e sulla funzione degli acidi nucleici e aminoacidi.")
60
Si tratta di un cofattore fondamentale nel metabolismo degli amminoacidi e acidi nucleici. Esso agisce come un donatore di atomo di carbonio. Si ottiene questo atomo di carbonio sequestrando formaldeide prodotta in altri processi .

61
INIBITORI DELLA SINTESI DEL ACIDO FOLICO
Antagonisti competitivi Antagonisti competitivi << >> La sulfanilamide compete con l’acido p-aminobenzoico legandosi al sito attivo dell’enzima diidropteroatosintetasi (DHPS) bloccando la sintesi dell’acido folico, precursore degli acidi nucleici
 bloccando la sintesi dell’acido folico, precursore degli acidi nucleici.")
62
Il Prontosil è stato il primo chemioterapico
I SULFAMIDICI sono stati i primi chemioterapici usati per inibire la crescita dei batteri. Il sulfamidico più semplice è la sulfanilamide un metabolita attivo del Prontosil Il Prontosil è stato il primo chemioterapico disponibile in commercio, sviluppato dai laboratori della Bayer in Germania I sulfamidici sono stati i primi chemioterapici usati per inibire la crescita dei batteri. Il sulfamidico più semplice è la sulfanilamide, che agisce come analogo dell’acido p-aminobenzoico, che, a sua volta, è parte della vitamina acido folico. La sulfanilamide compete con l’acido p-aminobenzoico legandosi al sito attivo dell’enzima diidropteroatosintetasi (DHPS) bloccando la sintesi dell’acido folico, precursore degli acidi nucleici. E’ attiva contro i batteri che sono in grado di sintetizzare acido folico, ma non contro gli organismi superiori, che devono procurarselo con la dieta. I sulfamidici hanno ampio spettro d’azione. Agiscono in quasi eguale misura sulla maggior parte dei batteri Gram(+) e Gram(-), alcuni protozoi, alcuni miceti e sulle clamidie. Sono stati utilizzati anche contro la malaria, la toxoplasmosi e la blastomicosi sudamericana. Non sono attivi contro gli actinomiceti, le spirochete e le ricketsie. Alcuni sulfamidici sono ancora farmaci di prima scelta, specialmente contro le infezioni delle vie urinarie, dell'intestino, dell'occhio, dell'orecchio e delle prime vie respiratorie. Non vengono usati per applicazioni topiche perchè hanno scarsa efficacia, contribuiscono alla formazione di ceppi resistenti e danno fenomeni di sensibilizzazione
 bloccando la sintesi dell’acido folico, precursore degli acidi nucleici. E’ attiva contro i batteri che sono in grado di sintetizzare acido folico, ma non contro gli organismi superiori, che devono procurarselo con la dieta. I sulfamidici hanno ampio spettro d’azione. Agiscono in quasi eguale misura sulla maggior parte dei batteri Gram(+) e Gram(-), alcuni protozoi, alcuni miceti e sulle clamidie. Sono stati utilizzati anche contro la malaria, la toxoplasmosi e la blastomicosi sudamericana. Non sono attivi contro gli actinomiceti, le spirochete e le ricketsie. Alcuni sulfamidici sono ancora farmaci di prima scelta, specialmente contro le infezioni delle vie urinarie, dell intestino, dell occhio, dell orecchio e delle prime vie respiratorie. Non vengono usati per applicazioni topiche perchè hanno scarsa efficacia, contribuiscono alla formazione di ceppi resistenti e danno fenomeni di sensibilizzazione.")
72
Ac. diidrofolico diidrofolato reduttasi (DHFR) TRIMETOPRIM
Il Trimetoprim è un antibiotico di sintesi appartenente alla categoria delle diaminopirimidine. Strutturalmente è una diaminopirimidina ,perciò presenterà analogie con il gruppo pteridinico del folato ( acido folico = anello pteridinico + PABA + acido glutammico ). Agisce come inibitore della diidrofolato reduttasi , impedendo così la sintesi dell'acido tetraidrofolico, che per il batterio è fondamentale. È infatti necessario per la sintesi di purine , pirimidine , timidine, metionina e quindi per la sintesi degli acidi nucleici dunque la replicazione. È un farmaco batteriostatico Il trimetoprim è analogo dell’acido diidrofolico. Esso esercita la sua azione inibente legandosi all’enzima diidrofolato riduttasi (DHFR), catalizzante la reazione che trasforma il diidrofolato in tetraidrofolato, cofattore richiesto per la sintesi delle purine, della timina, e di alcuni aminoacidi. Spettro d'azione e utilizzo [modifica] È simile a quello dei sulfamidici. Viene solitamente utilizzato in associazione con sulfametossazolo, che permette di inibire in sinergismo due tappe della sintesi dell'acido folico. Prende il nome di Cotrimoxazolo il sinergismo dei due antibiotici. È il farmaco di elezione per il trattamento di infezioni urinarie acute e croniche. Viene inoltre utilizzata questa associazione per il trattamento di infezioni causate da Pneumocystis jirovecii, alle vie respiratorie inferiori, per l'otite media e gonorree senza complicanze TRIMETOPRIM Ac. tetraidrofolico
. Agisce come inibitore della diidrofolato reduttasi , impedendo così la sintesi dell acido tetraidrofolico, che per il batterio è fondamentale. È infatti necessario per la sintesi di purine , pirimidine , timidine, metionina e quindi per la sintesi degli acidi nucleici dunque la replicazione. È un farmaco batteriostatico. Il trimetoprim è analogo dell’acido diidrofolico. Esso esercita la sua azione inibente legandosi all’enzima diidrofolato riduttasi (DHFR), catalizzante la reazione che trasforma il diidrofolato in tetraidrofolato, cofattore richiesto per la sintesi delle purine, della timina, e di alcuni aminoacidi. Spettro d azione e utilizzo [modifica] È simile a quello dei sulfamidici. Viene solitamente utilizzato in associazione con sulfametossazolo, che permette di inibire in sinergismo due tappe della sintesi dell acido folico. Prende il nome di Cotrimoxazolo il sinergismo dei due antibiotici. È il farmaco di elezione per il trattamento di infezioni urinarie acute e croniche. Viene inoltre utilizzata questa associazione per il trattamento di infezioni causate da Pneumocystis jirovecii, alle vie respiratorie inferiori, per l otite media e gonorree senza complicanze. TRIMETOPRIM. Ac. tetraidrofolico.")
73
INIBITORI DELLA SINTESI DEL ACIDO FOLICO
Antagonisti competitivi Antagonisti competitivi << >> La sulfanilamide compete con l’acido p-aminobenzoico legandosi al sito attivo dell’enzima diidropteroatosintetasi (DHPS) bloccando la sintesi dell’acido folico, precursore degli acidi nucleici
 bloccando la sintesi dell’acido folico, precursore degli acidi nucleici.")
74
ASSOCIAZIONE La conoscenza del processo biochimico ha reso possibile lo sviluppo di associazioni capaci di migliorare per sinergia l'efficacia dei farmaci: così inibitori di diversa natura (es. antifolico-sulfamidico) capaci di bloccare punti diversi dello stesso processo di formazione biologica (quello che porta alla formazione dell'acido tetraidrofolico) possono diventare inibitori per effetto sinergico. L'associazione di un sulfamidico e un inibitore della DHFR aumenta notevolmente l'attività antibatterica. Mentre il sulfamidico inibisce al livello della formazione dell'acido diidropteroico, l'antifolico inibisce al livello della riduzione dell'acido diidrofolico.
 capaci di bloccare punti diversi dello stesso processo di formazione biologica (quello che porta alla formazione dell acido tetraidrofolico) possono diventare inibitori per effetto sinergico. L associazione di un sulfamidico e un inibitore della DHFR aumenta notevolmente l attività antibatterica. Mentre il sulfamidico inibisce al livello della formazione dell acido diidropteroico, l antifolico inibisce al livello della riduzione dell acido diidrofolico.")
76
SOLFONI Sono strutturalmente correlati ai sulfamidici
DAPSONE ACEDAPSONE Profarmaco Come antibatterico viene utilizzato per combattere la lebbra causata dal Mycobacterium leprae. Viene saltuariamente utilizzato per prevenire infezioni causate da Pneumocystis pneumonia in pazienti immnunosoppressi o immnunodeficienti, o in quelle persone che non tollerano il trimetoprim ed il sulfametossazolo. Trova anche applicazioni nel trattamento della Porpora trombocitopenica idiopatica e della Malattia di Hailey-Hailey. Sono strutturalmente correlati ai sulfamidici. Dapsone o difenisolfone [4,4'diaminodifenilsolfonil-bisbenzenamino, 4,4'diaminodifenilsolfone o DDS]. Poco solubile in acqua, solubile negli acidi minerali diluiti, instabile alla luce. Ha basso costo, bassa tossicità, è farmaco di elezione per la sua discreta attività contro il Mycobacterium leprae, mentre ha debole attività contro il Micobacterium tuberculosis. E’ usato anche in dermatiti erpetiformi e in certe forme di malaria divenute resistenti ad antimalarici. La sua azione è potenziata dal trimetoprim. Inconvenienti: viene eliminato rapidamente, per cui deve essere somministrato a dosi elevate e per lunghi periodi, e ciò genera effetti tossici ed effetti secondari. Da qui è nata la necessità di avere derivati che in ambiente biologico cedessero lentamente il dapsone (acedapsone). Dapsone e il suo profarmaco è farmaco di elezione per la sua discreta attività contro il Mycobacterium leprae, mentre ha debole attività contro il Micobacterium tuberculosis. E’ usato anche in dermatiti erpetiformi e in certe forme di malaria divenute resistenti ad antimalarici. La sua azione è potenziata dal trimetoprim
. Dapsone e il suo profarmaco è farmaco di elezione per la sua discreta attività contro il Mycobacterium leprae, mentre ha debole attività contro il Micobacterium tuberculosis. E’ usato anche in dermatiti erpetiformi e in certe forme di malaria divenute resistenti ad antimalarici. La sua azione è potenziata dal trimetoprim.")
77
SOURCES OF ANTIBIOTICS
Examples molds penicillium penicillin cephalosporium cephalosporins actinomycetes tetracycline aminoglycosides (streptomycin) macrolides (erythromycin) chloramphenicol ivermectin rifamycins bacteria bacilli Dirt-dwelling organisms that form endospores and create antibiotics, possibly to deter bacterial competition. These organisms are unaffected by their own antibiotics, but can be susceptible to other antibiotics. Produce polypeptide antibiotics (e.g., polymyxin and bacitracin). B. cereus Zwittermicin synthetic oxazolidinones Linezolid (Zyvox)–Treat Gram-positive infections. Bind rRNA to prevent protein synthesis. Molds muffa
 macrolides (erythromycin) chloramphenicol ivermectin rifamycins. bacteria. bacilli. Dirt-dwelling organisms that form endospores and create antibiotics, possibly to deter bacterial competition. These organisms are unaffected by their own antibiotics, but can be susceptible to other antibiotics. Produce polypeptide antibiotics (e.g., polymyxin and bacitracin). B. cereus. Zwittermicin. synthetic. oxazolidinones. Linezolid (Zyvox)–Treat Gram-positive infections. Bind rRNA to prevent protein synthesis. Molds muffa.")
78
MECHANISM OF ACTION OF SELECTED ANTIBIOTICS
Inhibitors of cell wall synthesis Carbenicillin Inhibits transpeptidation enzymes. Activates lytic enzymes of cell wall. Pennicillin Inhibits transpeptidase enzymes. Activates lytic enzymes of cell wall. The affected bacterium will eventually lyse because the unsupported cell wall cannot withstand its growth. Vancomycin Inhibits transpeptidation in cross-linking peptidoglycans. Interferes with bacterial cells at many levels, disrupting cell wall synthesis, interfering with RNA, and damaging the plasma membrane. Inhibitors of nucleic acid synthesis Ciprofloxacin Inhibits DNA gyrase; interferes with DNA replication. Rifampin Blocks RNA synthesis by binding to and inhibiting RNA polymerase.

79
MECHANISM OF ACTION OF SELECTED ANTIBIOTICS
Inhibitors of protein synthesis Chloramphenicol Blocks formation of new peptide bonds during protein synthesis by binding to the 50S subunit of the ribosome. Erthromycin Binds the 50S subunit and blocks translocation of the new protein on the ribosome, thus effectively halting synthesis. Fusidic acid Blocks translocation. Linezolid Binds rRNA to prevent translation initiation and thus protein synthesis. Streptomycin Binds the 30S ribosomal subunit of the tuberculosis bacterium and prevents the ribosome from forming the complex necessary to initiate protein translation. Streptomycin is the first line of chemical defense against Mycobacterium tuberculosis. Tetracyclines Binds to the 30S subunit and blocks the addition of amino acids, producing incomplete and probably nonfunctional proteins. Metabolic inhibitors Dapsone Interferes with synthesis of folic acid, which is required for the synthesis of purines and thymidine and for the synthesis of the amino acids methionine and gycine. Sulfonamides Competitively inhibits dihydropteroate synthase, an enzyme that converts p-aminobenzoic acid (PABA) into folic acid. These drugs can also be incorporated into a compound that resembles dihydrofolate and that in turn can inhibit another enzyme in the pathway, dihydrofate reductase. Trimethoprim Inhibits dihydrofolate reductase, blocking tetrahydrofolate synthesis.
 into folic acid. These drugs can also be incorporated into a compound that resembles dihydrofolate and that in turn can inhibit another enzyme in the pathway, dihydrofate reductase. Trimethoprim. Inhibits dihydrofolate reductase, blocking tetrahydrofolate synthesis.")
80
Applicazioni terapeutiche degli antibiotici
INFEZIONI AD EZIOLOGIA SPECIFICA TRATTAMENTO Tifo Ciprofloxacina, Cloramfenicolo, Cotrimossazolo Sterilizzazione dei portatori di Salmonella typhi Ciprofloxacina Pefloxacina Brucellosi Minociclina + Streptomicina, Minociclina + Rifampicina, Cotrimossazolo +Streptomicina o Gentamicina Copertura antibiotica in corso di mononucleosi infettiva Macrolide Pertosse Eritromicina (o altri Macrolidi) oppure Cotrimossazolo Sifilide Penicillina G Benzatina, Ceftriaxone, Minociclina, Eritromicina Sifilide tardiva (da più di un anno o con interessamento cardiaco) Penicillina G Benzatina Minociclina Neurosifilide Penicillina G Benzatina Ceftriaxone Cloramfenicolo Sifilide congenita con liquor normale Penicillina G Benzatina,Eritromicina Sifilide congenita con liquor alterato Penicillina G Benzatina Eritromicina MENINGITI BATTERICHE Cefotaxima, Ceftriaxonesi ( Vancomicina con o senza Rifampicina per coprire gli Pneumococchi resistenti) nella meningiti da Streptococcus pneumoniae e Neisseria meningitidis LIinezolid nell’infezioni da aerobi gram-positivi, Stafilococco aureus e Pneumococco meticillino resistenti. Ceftazidima + Gentamicina meningite da Pseudomonas Ampicillina + Gentamicina meningite da Listeria monocitogenes oppure Cotrimossazolo Cefotaxima o Ceftriaxone Aztreonam per gli Enterobacilli gram-negativi
 oppure Cotrimossazolo. Sifilide. Penicillina G Benzatina, Ceftriaxone, Minociclina, Eritromicina. Sifilide tardiva (da più di un anno o con interessamento cardiaco) Penicillina G Benzatina. Minociclina. Neurosifilide. Penicillina G Benzatina Ceftriaxone Cloramfenicolo. Sifilide congenita con liquor normale. Penicillina G Benzatina,Eritromicina. Sifilide congenita con liquor alterato. Penicillina G Benzatina Eritromicina. MENINGITI BATTERICHE. Cefotaxima, Ceftriaxonesi ( Vancomicina. con o senza Rifampicina per coprire. gli Pneumococchi resistenti) nella meningiti da Streptococcus pneumoniae e Neisseria meningitidis. LIinezolid nell’infezioni da aerobi gram-positivi, Stafilococco aureus e Pneumococco meticillino resistenti. Ceftazidima + Gentamicina meningite da Pseudomonas. Ampicillina + Gentamicina meningite da Listeria monocitogenes oppure. Cotrimossazolo Cefotaxima o Ceftriaxone. Aztreonam per gli Enterobacilli gram-negativi.")
81
INFEZIONI EZIOLOGIA TRATTAMENTO ANTIBIOTICO
ENDOCARDITE Acuta in valvola nativa Entero-, Stafilo- e Streptococco Glicopeptide+ Aminoglicoside Teicoplanina, Vancomicina Gentamicina, Netilmicina, Ampicillina, Cefotaxima, Ceftazidima, Ceftriaxone Subacuta in valvola nativa Entero- e Streptococco Ampicillina + Aminoglicoside Valvola protesica Enterobacteriaceae e Stafilococco Glicopeptide + Aminoglicoside + Cefalosporina di 3° generazione Tossicomani Enterobacteriaceae e Stafilococco Aminoglicoside + Cefalosporina di 3a gener ARTRITI SETTICHE E OSTEOMIELITI Stafilococco aureo, Emofilo, Streptococco Stafilococco aureo, Neisseria gonorrhoeae Escherichia coli, Proteus, Pseudomonas Glicopeptide + Aminoglicoside + Metronidazolo o Clindamicina Glicopeptide + Piperacillina o Ceftazidima Ciprofloxacina o Levofloxacina +/- Rifampicina Teicoplanina, Gentamicina Netilmicina, Metronidazolo, Clindamicina, Piperacillina, Ceftazidima,Ciprofloxacina, Levofloxacina, Rifampicina

82
APPARATO RESPIRATORIO
INFEZIONI EZIOLOGIA TRATTAMENTO APPARATO RESPIRATORIO Faringo-tonsillite Streptococco Amoxicillina - Acido Clavulanico, o Cefixima, oClaritromicina, Penicillina G, Ceftriaxone Otite esterna Stafilococco aureus o Stafilococco epidermidis Ceftriaxone, Amoxicillina - Acido Clavulanico o Claritromicina ed altri macrolidi (Eritromicina, Azitromicina) Otite media e mastoidite Streptococcus pneumoniae 30-35%, Emofilo 20-25%, Moraxella catarrhalis 10-15% ; Stafilococco e Streptococco di gruppo A < 1% Amoxicillina - Acido Clavulanico o Claritromicina o altri macrolidi (Eritromicina, Azitromicina) Sinusiti Forme acute :.Streptococco, Emofilo ; in misura minore Stafilococco, Branhamella Forme croniche : Anaerobi oppure Levofloxacina, o Clindamicina o Cefotaxima Laringiti, Tracheiti e bronchiti acute Virale nel 90% dei casi, una modesta percentuale di casi è sostenuta da Micoplasmi, Bordetelle e Clamidie Amoxicillina - Acido Clavulanico Cefixima , Ciprofloxacina o Levofloxacina, o Claritromicina o altri macrolidi,oppure Cefotaxima
 Otite media e mastoidite. Streptococcus pneumoniae 30-35%, Emofilo 20-25%, Moraxella catarrhalis 10-15% ; Stafilococco e Streptococco di gruppo A < 1% Amoxicillina - Acido Clavulanico o Claritromicina o altri macrolidi (Eritromicina, Azitromicina) Sinusiti. Forme acute :.Streptococco, Emofilo ; in misura minore Stafilococco, Branhamella. Forme croniche : Anaerobi. oppure Levofloxacina, o. Clindamicina o Cefotaxima. Laringiti, Tracheiti e bronchiti acute. Virale nel 90% dei casi, una modesta percentuale di casi è sostenuta da Micoplasmi, Bordetelle e Clamidie. Amoxicillina - Acido Clavulanico. Cefixima , Ciprofloxacina o Levofloxacina, o Claritromicina o altri macrolidi,oppure Cefotaxima.")
83
APPARATO RESPIRATORIO Bronchite cronica riacutizzata
INFEZIONI EZIOLOGIA TRATTAMENTO APPARATO RESPIRATORIO Bronchite cronica riacutizzata Emofilo, Branhamella, Klebsiella ; più raramente Pneumococco, Stafilococco Levofloxacina Amoxicillina - Acido Clavulanico Broncopolmoniti e polmoniti Levofloxacina, Cefodizime oppure Amoxicillina - Acido Clavulanico, oppure Claritromicina Forme parenchimali interstiziali Virus, Micoplasmi, Clamidie, Legionelle, Rickettsie Stafilococchi meticillino-resistenti gram positivi meticillino-resistenti Betalattamina +Macrolide Monoterapia con Fluorochinoloni Teicoplanina o Vancomicina Tubercolosi Micobatteri tubercolari Isoniazide + Rifampicina Pirazinamide Rifampicina + Isoniazide + Pirazinamide+ Etambutolo + Streptomicina

84
INFEZIONI EZIOLOGIA TRATTAMENTO
TRATTO GASTROENTERICO Enterocolite e diarrea dei viaggiatori Escherichia coli, Shigelle, Salmonelle, Campilobacter jejuni, Yersinia enterocolitica Ciprofloxacina Cotrimossazolo Colecistiti e colangiti Escherichia coli, Klebsiella pneumoniae, Enterobacter, Citrobacter, Proteus, Morganella, Pseudomonas aeruginosa, Piperacillina – Tazobactam, Levofloxacina, Cefoperazone, Cefotaxima Carbapenemi (Imipenem e Meropenem) e l’Ampicillina in associazione a Gentamicina TRATTO GENITO-URINARIO Cistiti e cistopieliti Escherichia coli, Proteus, Pseudomonas aeruginosa, Klebsiella, Citrobacter, Serratia, ecc Levofloxacina o Amoxicillina - Acido Clavulanico o Netilmicina o Cefotaxima O Piperacillina - Tazobactam Pielonefriti acute Enterococco, gram-negativi Cefotaxima o Piperacillina - Tazobactam o Ciprofloxacina o Levofloxacina, Aztreonam o Cefodizime Prostatite acuta e cronica Escherichia coli, Klebsiella, Enterobacter, Proteus, Stafilococco aureus, Neisseria gonorrhoeae, Chlamydia trachomatis, Micoplasma, Enterococchi, Ciprofloxacina, Levofloxacina, Aztreonam, Minociclina, Cotrimossazolo. Claritromicina Malattia infiammatoria pelvica Neisseria gonorrhoeae, Streptococchi, Bacteroides fragilis, Escherichia coli, Klebsiella pneumoniae, Chlamydia trachomatis, Mycoplasma hominis Levofloxacina.+ Metronidazolo o Clindamicina Ceftriaxone + Minociclina Gonorrea Gonococco Uretriti non gonococciche Chlamydia trachomatis , Ureaplasma urealyticum. Claritromicina o Minociclina
 e l’Ampicillina in associazione a Gentamicina. TRATTO GENITO-URINARIO. Cistiti e cistopieliti. Escherichia coli, Proteus, Pseudomonas aeruginosa, Klebsiella, Citrobacter, Serratia, ecc. Levofloxacina o Amoxicillina - Acido Clavulanico o Netilmicina o Cefotaxima. O Piperacillina - Tazobactam. Pielonefriti acute. Enterococco, gram-negativi. Cefotaxima o Piperacillina - Tazobactam o Ciprofloxacina o Levofloxacina, Aztreonam o Cefodizime. Prostatite acuta e cronica. Escherichia coli, Klebsiella, Enterobacter, Proteus, Stafilococco aureus, Neisseria gonorrhoeae, Chlamydia trachomatis, Micoplasma, Enterococchi, Ciprofloxacina, Levofloxacina, Aztreonam, Minociclina, Cotrimossazolo. Claritromicina. Malattia infiammatoria pelvica. Neisseria gonorrhoeae, Streptococchi, Bacteroides fragilis, Escherichia coli, Klebsiella. pneumoniae, Chlamydia trachomatis, Mycoplasma hominis. Levofloxacina.+ Metronidazolo. o Clindamicina. Ceftriaxone + Minociclina. Gonorrea. Gonococco. Uretriti non gonococciche. Chlamydia trachomatis , Ureaplasma urealyticum. Claritromicina o Minociclina.")
85
PROFILASSI ANTIBIOTICA IN CHIRURGIA
INFEZIONI EZIOLOGIA TRATTAMENTO CUTANEE Foruncolo, ascesso, idrosadenite, impetigine, erisipela, fascite necrotizzante, morsi di animali Stafilococco aureo Streptococco piogene A Amoxicillina - Acido clavulanico, Ciprofloxacina, Levofloxacina Daptomicina Piede diabetico Anaerobi Imipenem e Meropenem associati a Metronidazolo o Clindamicina o, Piperacillina-Tazobactam, i Fluorochinoloni, o Teicoplanina PROFILASSI ANTIBIOTICA IN CHIRURGIA Chirurgia pulita (protesica e non) Stafilococco aureo, Stafilococchi coagulasi negativi, Enterobacteriaceae Teicoplanina o Vancomicina Chirurgia della testa e del collo Anaerobi del cavo orale, Gram-negativi aerobi (Enterobacteriaceae ed altri) Clindamicina + Gentamicina Chirurgia esofagea e gastroduodenale Enterobacteriaceae. Bacteroides spp. (escluso Bacteroides fragilis) Amoxicillina/acido clavulanico Ampicillina/sulbactam Chirurgia colorettale, appendicectomia Enterobacteriaceae, anaerobi (Bacteroides fragilis), Enterococchi Gentamicina + Metronidazolo Amoxicillina/acido clavulanico Ampicillina/sulbactam Piperacillina Chirurgia settica Enterobacteriaceae, anaerobi, Enterococchi Clindamicina+ Gentamicina Chirurgia biliare Enterobacteriaceae, Enterococcus spp Piperacillina Chirurgia urologica Enterobacteriaceae, Enterococchi Ciprofloxacina, Levofloxacina Chirurgia ostetrico-ginecologica Enterobacteriaceae, anaerobi (Bacteroides spp, Bacteroides fragilis), Streptococchi di gruppo B, Enterococchi Ceftizoxime, Ampicillina/sulbactam Taglio cesareo Enterobacteriaceae, anaerobi, enterococchi Chirurgia oftalmica Stafilococco epidermdis, Stafilococco aureo, Streptococchi, Enterobacteriaceae, Pseudomonas Gentamicina e Tobramicina + Ciprofloxacina o Levofloxacina Ferita traumatica Stafilococco aureo, Streptococchi di gruppo A, Clostridi Ampicillina/sulbactam
 Stafilococco aureo, Stafilococchi coagulasi negativi, Enterobacteriaceae. Teicoplanina o Vancomicina. Chirurgia della testa e del collo. Anaerobi del cavo orale, Gram-negativi aerobi (Enterobacteriaceae ed altri) Clindamicina + Gentamicina. Chirurgia esofagea e gastroduodenale. Enterobacteriaceae. Bacteroides spp. (escluso Bacteroides fragilis) Amoxicillina/acido clavulanico Ampicillina/sulbactam. Chirurgia colorettale, appendicectomia. Enterobacteriaceae, anaerobi (Bacteroides fragilis), Enterococchi. Gentamicina + Metronidazolo Amoxicillina/acido. clavulanico Ampicillina/sulbactam Piperacillina. Chirurgia settica. Enterobacteriaceae, anaerobi, Enterococchi. Clindamicina+ Gentamicina. Chirurgia biliare. Enterobacteriaceae, Enterococcus spp. Piperacillina. Chirurgia urologica. Enterobacteriaceae, Enterococchi. Ciprofloxacina, Levofloxacina. Chirurgia ostetrico-ginecologica. Enterobacteriaceae, anaerobi (Bacteroides spp, Bacteroides fragilis), Streptococchi di gruppo B, Enterococchi. Ceftizoxime, Ampicillina/sulbactam. Taglio cesareo. Enterobacteriaceae, anaerobi, enterococchi. Chirurgia oftalmica. Stafilococco epidermdis, Stafilococco aureo, Streptococchi, Enterobacteriaceae, Pseudomonas. Gentamicina e Tobramicina + Ciprofloxacina o Levofloxacina. Ferita traumatica. Stafilococco aureo, Streptococchi di gruppo A, Clostridi. Ampicillina/sulbactam.")
Presentazioni simili

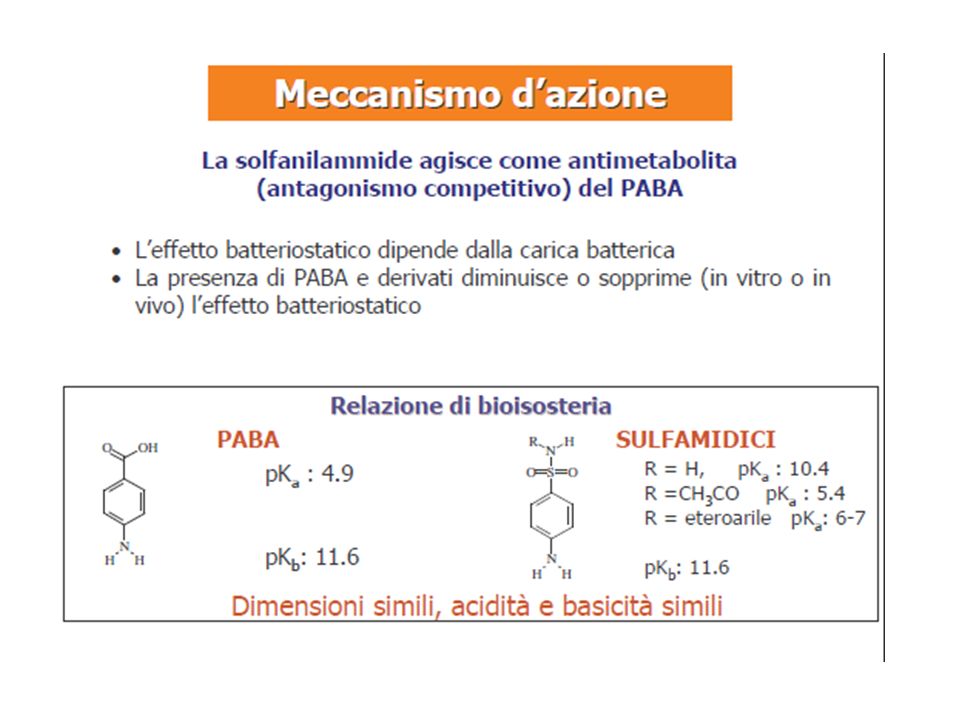

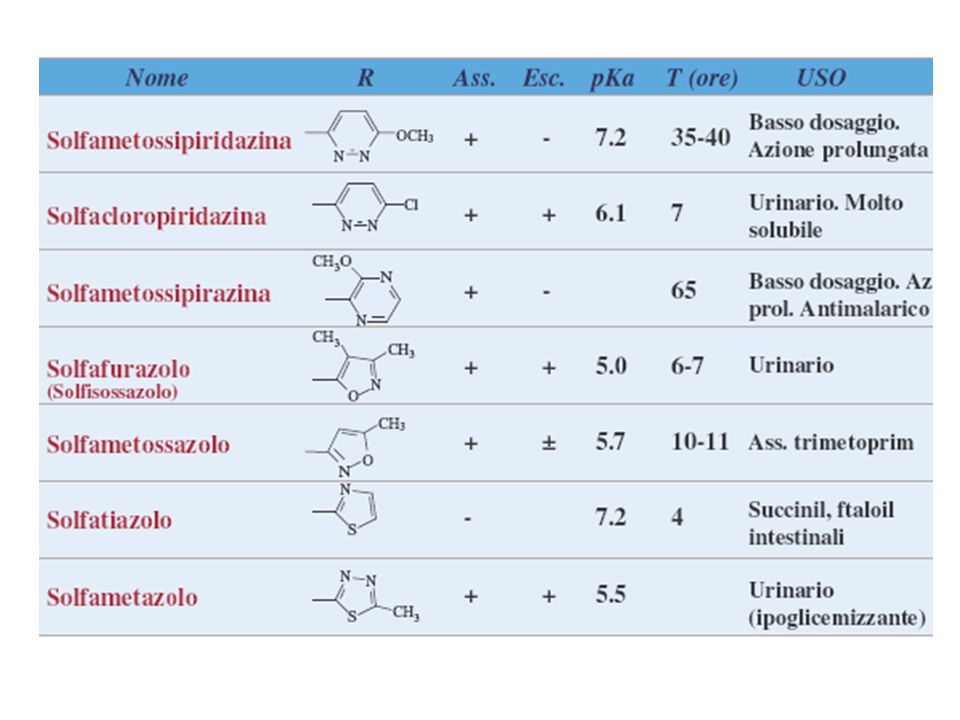
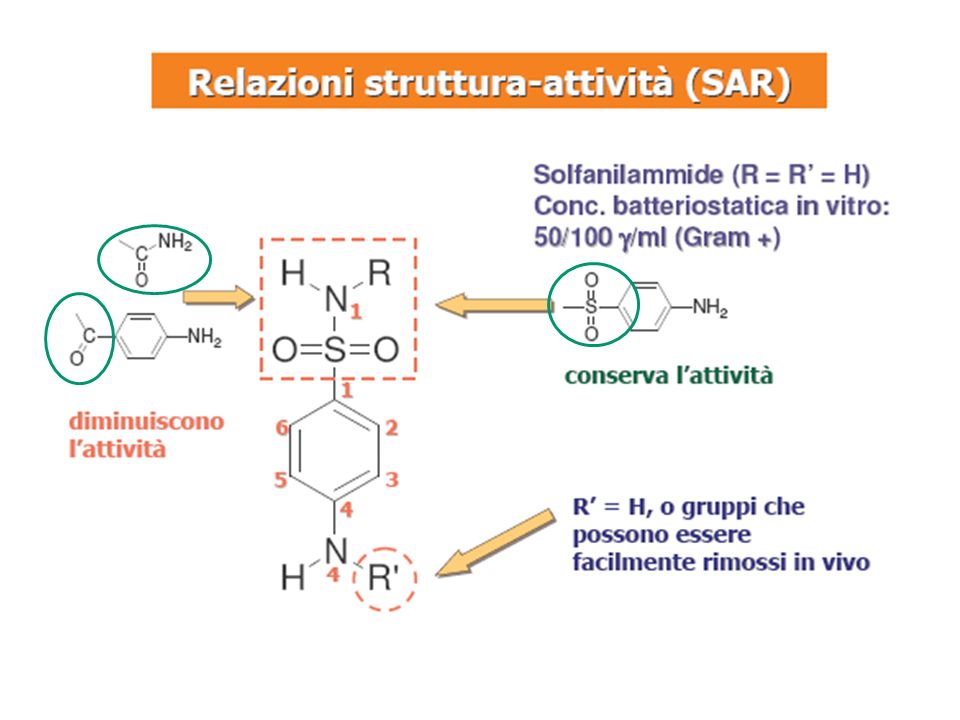

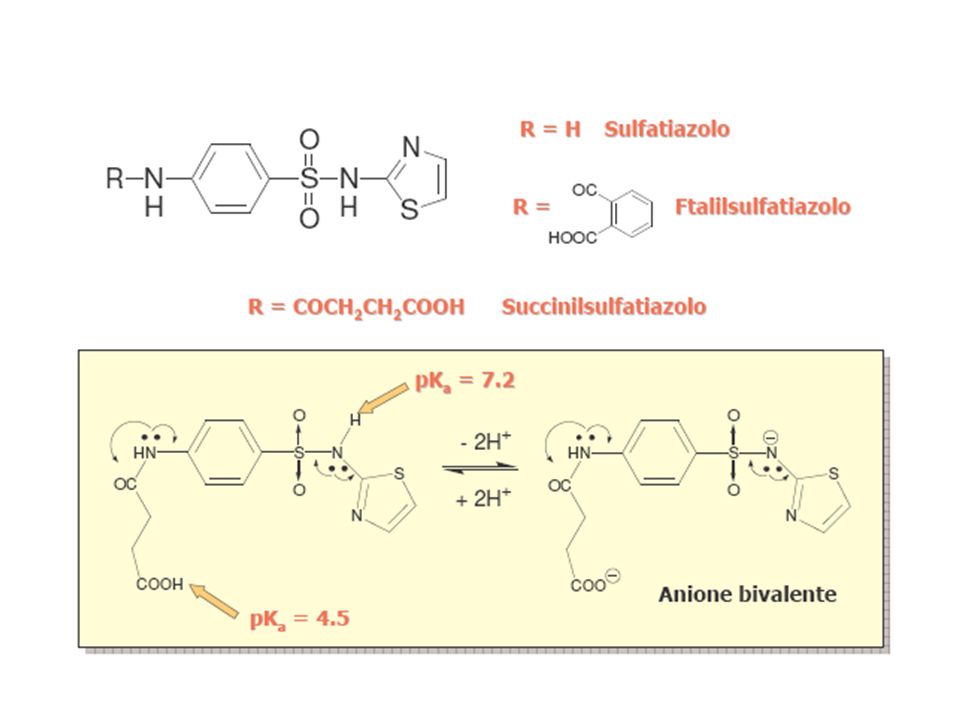
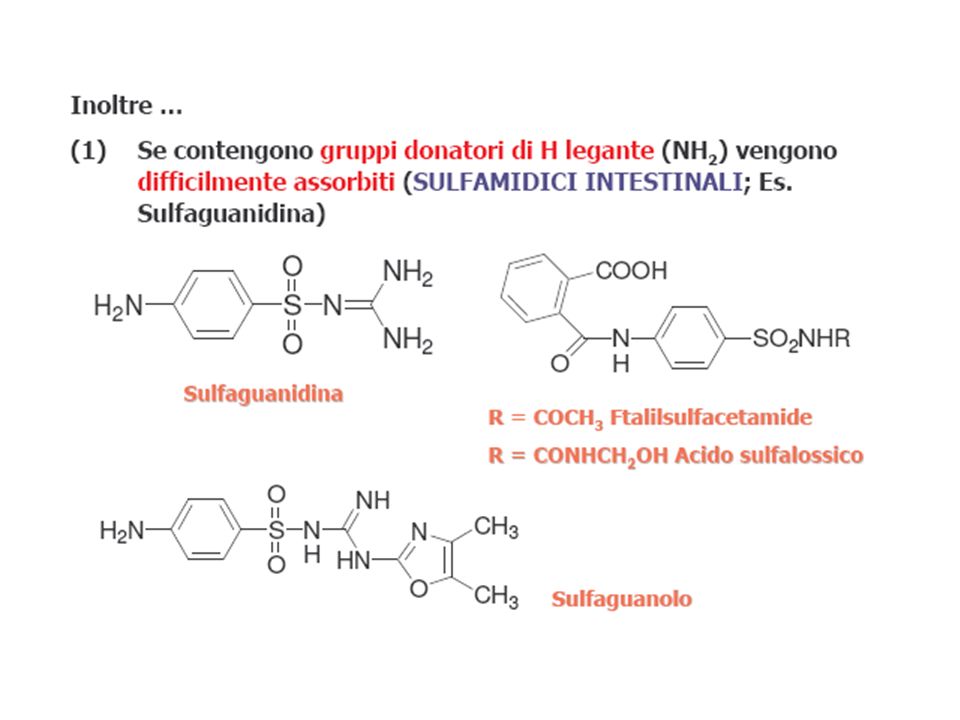
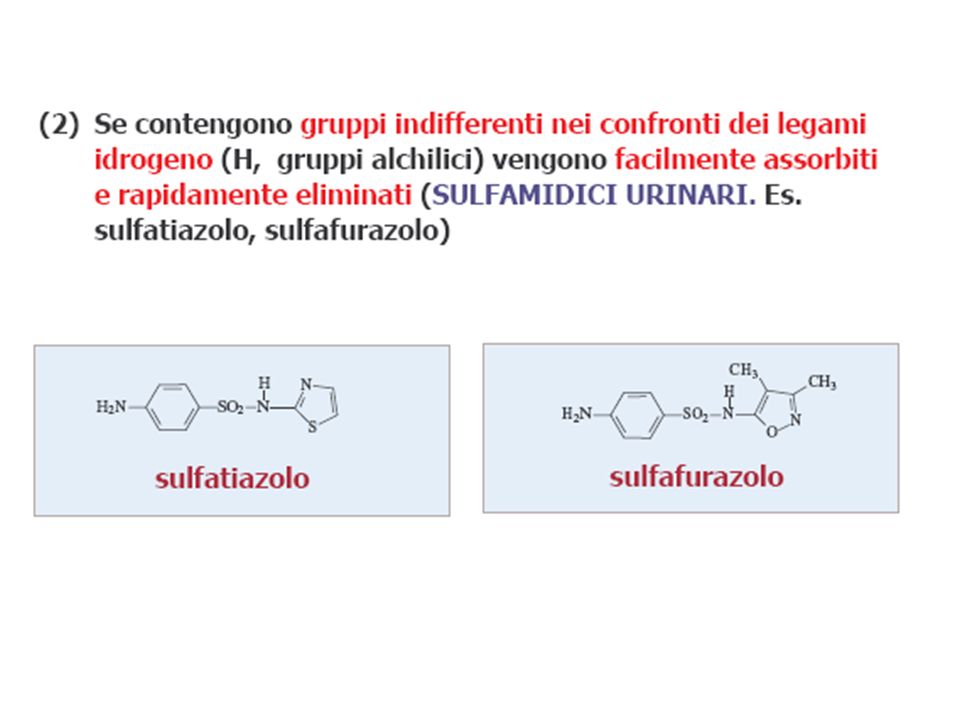
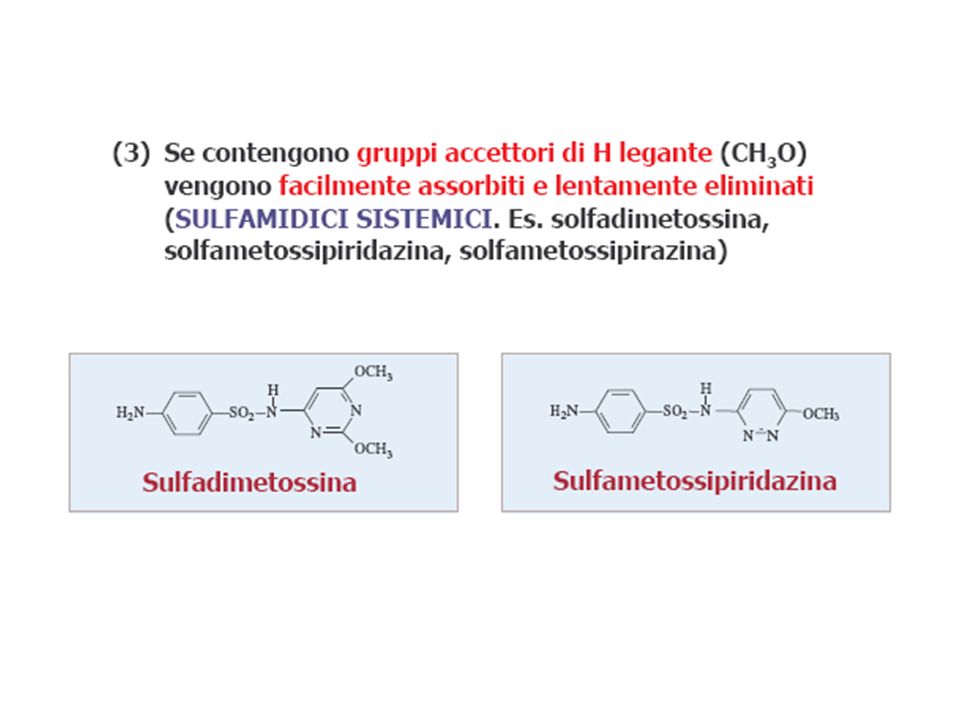
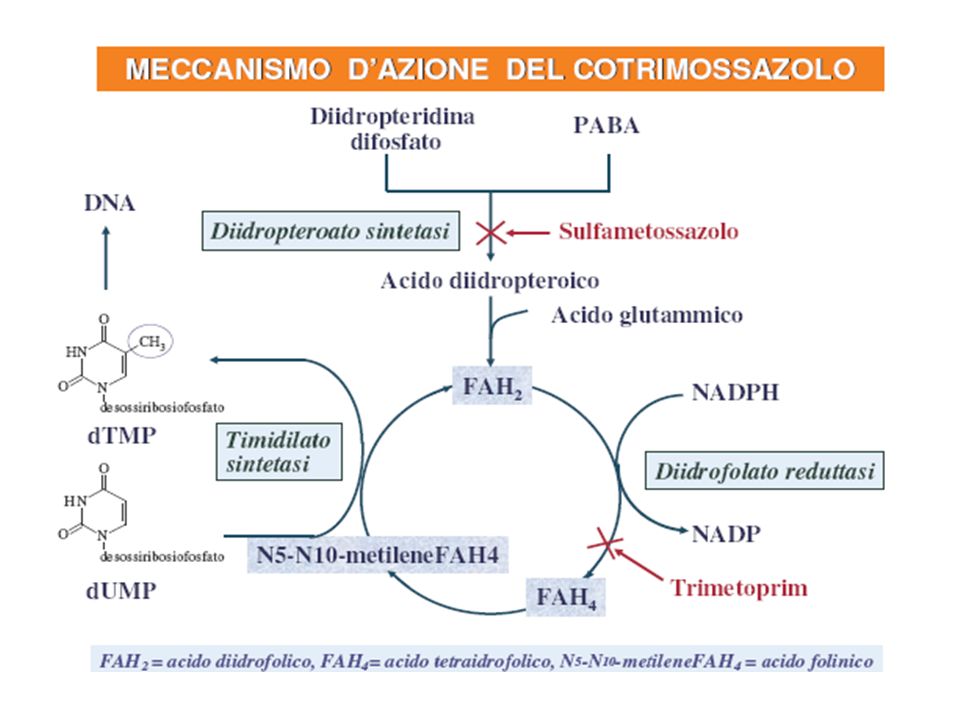














>")




