Scaricare la presentazione
1
Cromatografia

2
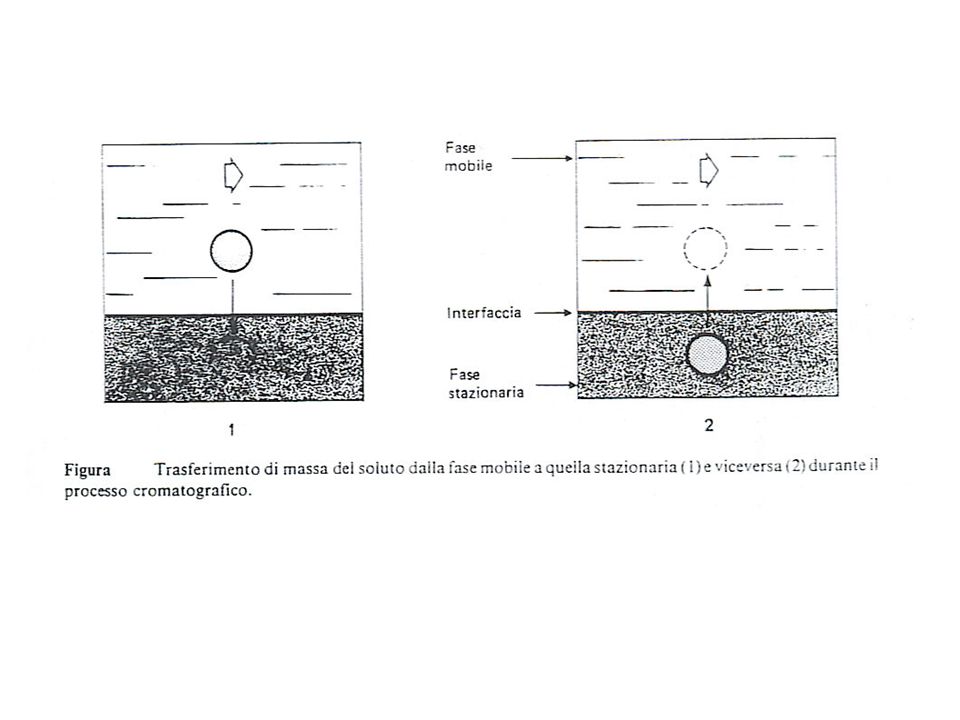
Con il termine cromatografia si intende un insieme di tecniche di separazione e purificazione di miscele di soluti, che presentano alcune caratteristiche fondamentali in comune: 1) La separazione è basata sulla distribuzione dei soluti tra due fasi, di cui una fissa (fase stazionaria) e l’ altra in movimento (fase mobile) 2)La fase fissa, solida o liquida che sia, è un sistema ad elevato sviluppo superficiale, attraverso il quale fluisce la fase mobile Alcuni soluti tendono a rimanere di più di altri in fase fissa e quindi vengono selettivamente ritardati: I meccanismi che introducono il ritardo selettivo sono essenzialmente: Adsorbimento ripartizione esclusione scambio ionico affinità Il risultato è la produzione di bande ad elevata concentrazione dei componenti separati. Indipendentemente dal meccanismo effettivo della separazione, legato al tipo di interazione fra soluto e fase stazionaria. La separazione cromatografica è un processo dinamico nel quale l’ analita si trasferisce dalla fase mobile a quella stazionaria, e viceversa.
 La separazione è basata sulla distribuzione dei soluti tra due fasi, di cui una fissa (fase stazionaria) e l’ altra in movimento (fase mobile) 2)La fase fissa, solida o liquida che sia, è un sistema ad elevato sviluppo superficiale, attraverso il quale fluisce la fase mobile. Alcuni soluti tendono a rimanere di più di altri in fase fissa e quindi vengono selettivamente ritardati: I meccanismi che introducono il ritardo selettivo sono essenzialmente: Adsorbimento. ripartizione. esclusione. scambio ionico. affinità. Il risultato è la produzione di bande ad elevata concentrazione dei componenti separati. Indipendentemente dal meccanismo effettivo della separazione, legato al tipo di interazione fra soluto e fase stazionaria. La separazione cromatografica è un processo dinamico nel quale l’ analita si trasferisce dalla fase mobile a quella stazionaria, e viceversa.")
3
In chimica e nelle scienze farmaceutiche, il coefficiente di ripartizione (o coefficiente di distribuzione) rappresenta il rapporto tra le concentrazioni di un composto all'interno delle due fasi di una miscela di due liquidi immiscibili all'equilibrio. L'importanza di questo coefficiente consiste nell'indicazione del livello di idrofilia o idrofobia di una sostanza chimica, log P= Log ([A]olio / [A]acqua) È il logaritmo del rapporto tra la concentrazione del soluto nella fase organica e quella nella fase acquosa.
 È il logaritmo del rapporto tra la concentrazione del soluto nella fase organica e quella nella fase acquosa.")
5
Classificazione delle tecniche cromatografiche
Esiste una enorme varietà di tecniche cromatografiche, che differiscono per la natura dei processi coinvolti e per le apparecchiature impiegate: Per una classificazione di queste tecniche si fa generalmente riferimento a: natura delle fasi meccanismo di distribuzione dispositivo che contiene la fase stazionaria metodo di esecuzione in base alla natura delle fasi (Fissa o mobile) si distinguono due branche fondamentali: la cromatografia a fase mobile liquida (LC) la cromatografia a fase mobile gassosa (GC)
 si distinguono due branche fondamentali: la cromatografia a fase mobile liquida (LC) la cromatografia a fase mobile gassosa (GC)")
6
A seconda della natura del processo di distribuzione implicato, si parla di:
cromatografia di adsorbimento (a fase stazionaria solida) cromatografia di ripartizione (a fase stazionaria liquida). Mentre la cromatografia di adsorbimento è solitamente impiegata per purificare composti su scala preparativa o semipreparativa, quella di ripartizione è più versatile e si presta anche e soprattutto ad applicazioni analitiche. Essa ha avuto un notevole sviluppo con la cromatografia gas-liquido (GS-L) e la cromatografia liquida ad elevate prestazioni HPLC (liquido-liquido). Le due tecniche sono complementari, la GC essendo adatta per la separazione di molecole non polari relativamente piccole e termostabili, l’ HPLC al contrario essendo indicata per grosse molecole polari anche termolabili.
 cromatografia di ripartizione (a fase stazionaria liquida). Mentre la cromatografia di adsorbimento è solitamente impiegata per purificare composti su scala preparativa o semipreparativa, quella di ripartizione è più versatile e si presta anche e soprattutto ad applicazioni analitiche. Essa ha avuto un notevole sviluppo con la cromatografia gas-liquido (GS-L) e la cromatografia liquida ad elevate prestazioni HPLC (liquido-liquido). Le due tecniche sono complementari, la GC essendo adatta per la separazione di molecole non polari relativamente piccole e termostabili, l’ HPLC al contrario essendo indicata per grosse molecole polari anche termolabili.")
7
Se si prende come criterio di suddivisione il tipo di dispositivo in cui si realizza il contatto tra la fase mobile e fase stazionaria, si distingue fra: cromatografia su colonna cromatografia planare Nel primo tipo la fase mobile fluisce per gravità o sotto pressione in un tubo lungo e stretto di vetro, metallo o polimero plastico. Nella cromatografia planare la fase mobile scorre per capillarità sulla fase stazionaria disposta su di una superficie piana (cromatografia su strato sottile) o negli interstizi di un foglio di carta per cromatografia (cromatografia su carta). La gas cromatografia può essere ovviamente effettuata solo su colonna.
 o negli interstizi di un foglio di carta per cromatografia (cromatografia su carta). La gas cromatografia può essere ovviamente effettuata solo su colonna.")
8
Meccanismi di ritardo operanti in cromatografia
I meccanismi che introducono il ritardo selettivo sono: Adsorbimento Ripartizione Scambio ionico Esclusione Affinità

9
Adsorbimento La fase stazionaria è un solido sulla cui superficie si trovano dei siti attivi in grado di stabilire legami secondari con le molecole degli analiti. Questi si distribuiranno tra le due fasi (sciogliendosi in quella mobile ed adsorbendosi alla superficie di quella fissa in un equilibrio continuo) in modo differente a seconda delle affinità relative.
 in modo differente a seconda delle affinità relative.")
10
Le proprietà importanti della fase stazionaria (adsorbente) sono:
Insolubilità nei solventi impiegati Inerzia chimica verso gli analiti ed i solventi Elevato sviluppo superficiale per unità di peso (in m2/g); Si hanno quindi migliori separazioni con fasi stazionarie ad elevato rapporto superficie-peso. Porosità (volume totale dei pori per grammo, in cm3/g). Dimensioni delle particelle; minore è il diametro delle particelle, più veloce è il trasferimento di massa e migliore l’ efficienza; il limite è costituito dalla resistenza al flusso della fase mobile. Attività superficiale o potere adsorbente: è il complesso delle forze di attrazione che l’ adsorbente esercita sul soluto: interazioni dipolo-dipolo Forze di induzione Forze di dispersione Interazioni per trasferimento di carica Legame H Legame ionico o covalente
; Si hanno quindi migliori separazioni con fasi stazionarie ad elevato rapporto superficie-peso. Porosità (volume totale dei pori per grammo, in cm3/g). Dimensioni delle particelle; minore è il diametro delle particelle, più veloce è il trasferimento di massa e migliore l’ efficienza; il limite è costituito dalla resistenza al flusso della fase mobile. Attività superficiale o potere adsorbente: è il complesso delle forze di attrazione che l’ adsorbente esercita sul soluto: interazioni dipolo-dipolo. Forze di induzione. Forze di dispersione. Interazioni per trasferimento di carica. Legame H. Legame ionico o covalente.")
11
Ad esclusione delle interazioni che corrispondono a veri e propri legami chimici (chemiosorbimento), nelle rimanenti, le energie in gioco sono ridotte al punto da garantire la reversibilità degli equilibri di adsorbimento. Le forze di dispersione sono di natura aspecifica e sono sempre presenti. Il contributo relativo delle forze di dispersione all’ energia totale di adsorbimento varia dal 100% degli idrocarburi saturi a meno del 50% per molecole polari come l’ acetone o l’ alcool. Il principale meccanismo di adsorbimento (prevalentemente nel caso che l’ adsorbente sia silice) è l’ interazione via legame idrogeno degli ossidrili superficiali con i gruppi funzionali dei soluti. Gli adsorbenti più comunemente usati sono costituiti (ad eccezione del carbone attivo) da sostanze ossigenate per lo più inorganiche.
 da sostanze ossigenate per lo più inorganiche.")
12
2Na2O . SiO2 + 2H2SO4 H4SiO4 + 2 Na2SO4
Gel di silice E’ il materiale di maggior uso. Costituito da acido silicico amorfo e altamente poroso, viene fatto precipitare, sotto forma di particelle dure e leggermente opache, trattando con acido solforico il vetro solubile (silicato di sodio). 2Na2O . SiO2 + 2H2SO4 H4SiO4 + 2 Na2SO4 La polimerizzazione dell’ acido silicico, con eliminazione di acqua, porta alla formazione di granuli porosi sulla cui superficie sono presenti i centri attivi Si-OH-, i silanoli. Allumina (ossidi di alluminio) Questo materiale viene preparato a partire da idrossidi di alluminio naturali (come ì’ idroargillite). Mediante opportuni trattamenti e una forte disidratazione a °C, si può ottenere allumina in tre forme, acida, basica, e neutra.
. 2Na2O . SiO2 + 2H2SO4 H4SiO4 + 2 Na2SO4. La polimerizzazione dell’ acido silicico, con eliminazione di acqua, porta alla formazione di granuli porosi sulla cui superficie sono presenti i centri attivi Si-OH-, i silanoli. Allumina (ossidi di alluminio) Questo materiale viene preparato a partire da idrossidi di alluminio naturali (come ì’ idroargillite). Mediante opportuni trattamenti e una forte disidratazione a °C, si può ottenere allumina in tre forme, acida, basica, e neutra.")
13
L’ attività degli adsorbenti può essere diminuita, mediante aggiunta di saturatori (acqua per quelli polari) od aumentata mediante riscaldamento. Comunemente vengono impiegati adsorbenti polari ed eluenti poco polari. Gli equilibri di adsorbimento sono processi competitivi, in cui le molecole di eluente competono con i soluti per i siti attivi della fase stazionaria. I solventi molto polari sono dotati di potere eluente maggiore di quelli poco polari. Gli eluenti sono ordinati in serie a potere eluente crescente (serie eluotrope), utili per programmare le eluizioni od effettuare i gradienti. Il potere eluente di un solvente è legato alla sua costante dielettrica (e0). (imparare a memoria la serie eluotropa di Trappe)
, utili per programmare le eluizioni od effettuare i gradienti. Il potere eluente di un solvente è legato alla sua costante dielettrica (e0). (imparare a memoria la serie eluotropa di Trappe).")
14
La valutazione dei parametri relativi ai soluti che influiscono sull’ adsorbimento in un dato sistema cromatografico non è semplice, entrano in gioco, oltre alle energie di adsorbimento dei gruppi funzionali presenti nel soluto: le dimensioni molecolari le interazioni fra molecole di soluto, il volume occupato da uno strato monomolecolare di soluto il valore di costante dielettrica L’ ordine decrescente con cui le varie classi di composti organici vengono trattenute da una fase stazionaria polare come l’ allumina è il seguente: Acidi e basi > alcooli e mercaptani>aldeidi e chetoni> esteri ed alogenuri> idrocarburi aromatici > alcheni ed alchini > alcani Nel caso dei composti di natura aromatica: -COOH > -CONH2 > -OH >- NH2 - > NHAc- >-COOCH3 > N(CH3)2 >- OBz >- NO2 > - OCH3 <- H
2 >- OBz >- NO2 > - OCH3 <- H.")
15
Esecuzione pratica di una cromatografia su colonna:
Il riempimento della colonna con la fase stazionaria solida va fatto in modo tale da ottenere una stratificazione regolare e omogenea dell’ adsorbente che costituisce la fase mobile. Quando si usa l’ allumina o il gel di silice, si sistema prima un po’ di cotone nella parte inferiore della colonna (vicino al rubinetto) poi si aggiunge il solvente, quindi un po’ di sabbia; e infine lentamente e in maniera continua l’ allumina evitando la formazione di crepe o bolle (per questo si può battere la colonna con un martelletto di gomma). Quando si usa la silice si agita l’ adsorbente insieme al solvente in un becher in modo da far svolgere le bollicine di aria trattenute dalla silice, quindi si versa la sospensione così ottenuta in colonna, dove precedentemente si era sistemato il cotone e la sabbia, che hanno la funzione di impedire che l’ adsorbente esca dal rubinetto della colonna; questa funzione può essere svolta da un setto di vetro poroso. Prima di introdurre la soluzione della miscela di sostanze da cromatografare, si fa scorrere aprendo il rubinetto l’ eccesso di solvente fino a quando il suo livello ha raggiunto il livello superiore dell’ adsorbente che si è stratificato nella colonna. E’ bene aggiungere, prima di quest’ ultima operazione un po’ di sabbia e un batuffolo di cotone idrofilo che hanno la funzione di proteggere la regolarità della superficie dell’ adsorbente dalle successive aggiunte di solvente (eluente).
 poi si aggiunge il solvente, quindi un po’ di sabbia; e infine lentamente e in maniera continua l’ allumina evitando la formazione di crepe o bolle (per questo si può battere la colonna con un martelletto di gomma). Quando si usa la silice si agita l’ adsorbente insieme al solvente in un becher in modo da far svolgere le bollicine di aria trattenute dalla silice, quindi si versa la sospensione così ottenuta in colonna, dove precedentemente si era sistemato il cotone e la sabbia, che hanno la funzione di impedire che l’ adsorbente esca dal rubinetto della colonna; questa funzione può essere svolta da un setto di vetro poroso. Prima di introdurre la soluzione della miscela di sostanze da cromatografare, si fa scorrere aprendo il rubinetto l’ eccesso di solvente fino a quando il suo livello ha raggiunto il livello superiore dell’ adsorbente che si è stratificato nella colonna. E’ bene aggiungere, prima di quest’ ultima operazione un po’ di sabbia e un batuffolo di cotone idrofilo che hanno la funzione di proteggere la regolarità della superficie dell’ adsorbente dalle successive aggiunte di solvente (eluente).")
16
La scelta dell’ adsorbente, del rapporto peso adsorbente / peso miscela da cromatografare, e infine dei solventi con cui eluire i componenti della miscela, va fatta in base sia alla natura delle sostanze sia alla difficoltà di separazione delle stesse. Maggiore sarà la difficoltà di separazione più alto sarà il rapporto adsorbente/miscela necessario per ottenere una soddisfacente separazione. Nell’ analisi per eluizione che è la tecnica di lavoro più comunemente usata dopo aver effettuato il riempimento della colonna nelle modalità prima descritte, si fa adsorbire sulla parte superiore della colonna la soluzione della miscela nella minima quantità di solvente che in generale è lo stesso con cui si è effettuato il riempimento; a questo punto si comincia ad eluire con il solvente che si può cambiare nel corso della cromatografia. A causa delle diverse affinità delle sostanze componenti della miscela, per la fase fissa e per quella mobile si avrà che un composto verrà ritardato rispetto all’ altro e quindi uscirà dalla colonna in un tempo diverso. Si raccolgono in palloncini di piccole dimensioni le frazioni di eluato che escono dal rubinetto, e poi si evapora il solvente.

17
Non sempre si ha una separazione netta, e spesso alcune frazioni contengono una miscela di sostanze che si può sottoporre ad un ulteriore processo di purificazione. La figura seguente mostra il susseguirsi di alcune fasi della separazione cromatografica di una miscela di due sostanze.

18
Rappresentazione Grafica di una separazione cromatografica ( curva di eluizione o cromatogramma)
Per seguire l’ andamento di una cromatografia si può riportare in un diagramma cartesiano la concentrazione della soluzione all’ uscita della colonna CL in funzione del volume di solvente eluito VL; si ottengono così delle curve di eluizione, caratteristiche per sistemi che si considerano e delle tecniche impiegate. Figura 1

19
Isoterma di assorbimento lineare
Ammettendo che manchino sia fenomeni di diffusione nell’ eluente, che irregolarità di riempimento della colonna e che l’ equilibrio di distribuzione tra le due fasi si stabilisca istantaneamente, la sostanza dovrebbe migrare attraverso l’ adsorbente mantenendosi in una zona nettamente delimitata, all’ interno della quale è verificata la relazione Cf/Cm=K; la sostanza dovrebbe comparire nell’ eluato dando luogo a una curva di eluizione simmetrica ed a concentrazione costante, come mostrato in fig. A Questo caso di può indicare come lineare-ideale. In pratica le condizioni ideali non si realizzano mai completamente, e le sostanze escono dalla colonna in maniera non lineare con variazioni più o meno nette, di solito si nota allargamento della banda con elevata scodatura.

20
In Fig 1 è rappresentato un cromatogramma ideale; l’ equilibrio viene raggiunto istantaneamente, non ci sono fenomeni di diffusione e le isoterme sono lineari. Nella colonna sono introdotte due sostanze A e B. Le bande di A e di B non cambiano forma. Con il simbolo Rf si indica il seguente rapporto: Rf = Distanza percorsa dal soluto/ distanza percorsa dal fronte dell’ eluente. . I valori di Rf come definiti, sono caratteristici e riproducibili per ciascun soluto in un dato sistema adsorbente-eluente, essi dipendono oltre che dalla temperatura, dalla concentrazione del soluto e dalla velocità di flusso dell’ eluente. fronte Distanza percorsa dal soluto A Distanza percorsa dal soluto B

21
Ripartizione Se la fase stazionaria è un liquido, il meccanismo che determina la separazione è la ripartizione delle sostanze tra le due fasi liquide (immiscibili tra loro) in base alle affinità relative; la velocità di migrazione dei soluti con la fase mobile è determinata dai rispettivi coefficienti di ripartizione (K.
 in base alle affinità relative; la velocità di migrazione dei soluti con la fase mobile è determinata dai rispettivi coefficienti di ripartizione (K.")
22
In un sistema cromatografico liquido-liquido la fase stazionaria è costituita da un liquido supportato su materiale granulare finemente suddiviso; ogni granulo viene ad essere rivestito in modo abbastanza uniforme da una sottile pellicola di liquido, realizzando così una elevata superficie di contatto per lo scambio con la fase mobile. I vantaggi che la ripartizione offre rispetto all’ adsorbimento sono: isoterme lineari ed assenza di alterazioni nel caso di soluti polari labili.

23
- Celite / acqua (od altri solventi) Cellulosa /acqua
I solidi di supporto dovrebbero essere inerti ed agire solo meccanicamente; in pratica c’e’ sempre una componente di adsorbimento. Le combinazioni più comuni sono: Fase diretta - Gel di silice / acqua adsorbita su di essa - Celite / acqua (od altri solventi) Cellulosa /acqua Si può verificare che la fase mobile, per azione meccanica, trascini via la fase stazionaria (o la silice impregnata di acqua) dal supporto; per questo la fase stazionaria non deve essere molto volatile. Per ovviare a questo inconveniente (stripping) si ricorre alla saturazione della fase mobile con la fase stazionaria, oppure all’ uso di fasi fisse legate chimicamente al supporto (cromatografia a fasi legate, BPC)
 Cellulosa /acqua. Si può verificare che la fase mobile, per azione meccanica, trascini via la fase stazionaria (o la silice impregnata di acqua) dal supporto; per questo la fase stazionaria non deve essere molto volatile. Per ovviare a questo inconveniente (stripping) si ricorre alla saturazione della fase mobile con la fase stazionaria, oppure all’ uso di fasi fisse legate chimicamente al supporto (cromatografia a fasi legate, BPC)")
24
Fasi invertite Si parla di cromatografia a fase normale quando la fase stazionaria è più polare di quella mobile; l’ opposto si verifica nella cromatografia a fase inversa. In generale la fase stazionaria è polare a causa delle caratteristiche dei supporti solidi. Può essere conveniente invece di disporre di una fase fissa apolare ed una fase mobile polare. Uno dei processi più comuni per invertire la polarità dei supporti quali silice o celite è la sililazione: per trattamento con dimetilclorosilano la superficie dei solidi, che era prima ricoperta da gruppi ossidrilici, espone dei gruppi metilici o a lunga catena idrocarburica. Fasi legate comuni portano catene C8 e C12 lineari

25
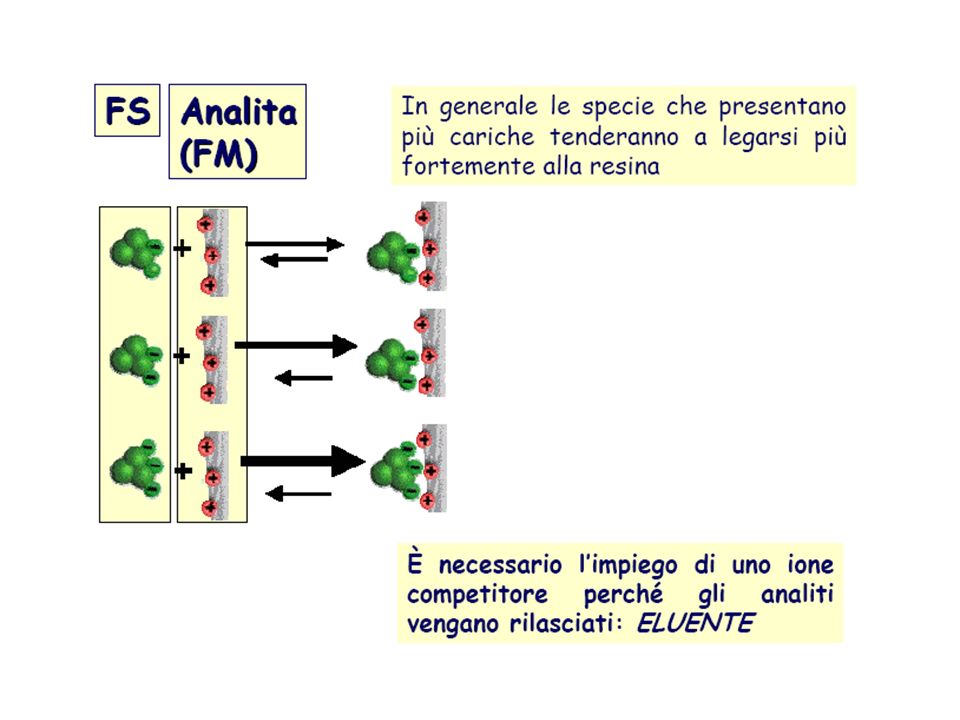
Cromatografia a scambio ionico:
E` una tecnica usata come cromatografia su carta, su colonna e anche per la cromatografia ad alta pressione (HPLC). La distribuzione del soluto fra fase fissa e fase mobile avviene mediante scambi di natura chimica e non di natura fisica. Per ottenere questo tipo di interazioni vengono utilizzate delle resine a scambio ionico. Queste sono costituite da un polimero base, formato da lunghe catene lineari di stirene unite tra di loro da molecole di di-vinil benzene che formano un ponte. L’ anello aromatico dello stirene lega gruppi di natura acida o basica. Stirene: Divinilbenzene:
. La distribuzione del soluto fra fase fissa e fase mobile avviene mediante scambi di natura chimica e non di natura fisica. Per ottenere questo tipo di interazioni vengono utilizzate delle resine a scambio ionico. Queste sono costituite da un polimero base, formato da lunghe catene lineari di stirene unite tra di loro da molecole di di-vinil benzene che formano un ponte. L’ anello aromatico dello stirene lega gruppi di natura acida o basica. Stirene: Divinilbenzene:")
26
Un polimero può essere così schematizzato:
A seconda della natura del gruppo polare, le resine possono essere suddivise in cationiche o anioniche. Le prime contengono un gruppo di natura acida, le seconde un gruppo di natura basica. I gruppi acidi sono: - SO3H (acide forti) COOH acide, OH debolmente acide. I gruppi basici sono : N(CH3)3 basiche forti, NH+(CH3)2 (basiche); NH2++(CH3) basiche deboli
 COOH acide, OH debolmente acide. I gruppi basici sono : N(CH3)3 basiche forti, NH+(CH3)2 (basiche); NH2++(CH3) basiche deboli.")
27
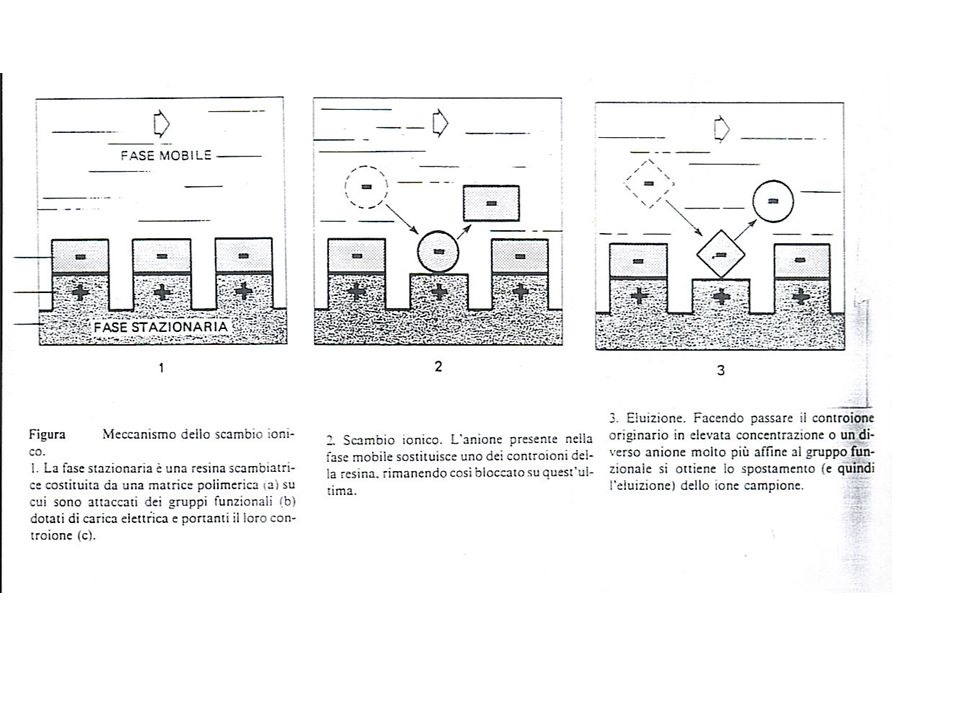
Resina-SO3H + Na+ resina-SO3Na + H+
Queste resine sono capaci di scambiare i loro ioni con quelli della soluzione con cui vengono a contatto. Per esempio la resina solfonica scambia lo ione H+ con lo ione Metallico+ presente in soluzione. Es il sodio+ Es: Resina-SO3H + Na+ resina-SO3Na + H+ Lo ione Na+ può essere portato via dalla soluzione per scambio con il protone.Ma la soluzione rimane acida. E’ una tecnica usata, per esempio, negli impianti di produzione di acqua demineralizzata. Dove l’ acua passa per una resina acida e poi per una basica, in modo alla fine da uscire a pH 7

28
Una resina è caratterizzata da 2 parametri:
le dimensioni dei pori della struttura polimerica e la capacità di scambiare ioni, espressa in n. eq. per grammo di resina secca. La percentuale di divinilbenzene determina il diametro dei pori della struttura a rete del polimero: maggiore è la percentuale e più piccolo è il diametro dei pori della struttura, maggiore la sua selettività. La larghezza dei pori è misurata in mesh, maggiore è il numero di mesh, minore il numero di pori. Consideriamo lo scambio tra 2 ioni Ar (ione legato alla resina) e Bs (ione in soluzione)
 e Bs (ione in soluzione)")
29
K= [As]*[Br]/[Ar] * [Bs]
Si stabilirà il seguente equilibrio: Ar + Bs As + Br Questo equilibrio è regolato dalla legge di azione di massa: K= [As]*[Br]/[Ar] * [Bs] K è il coefficiente di selettività. Esso è una misura dell’ affinità relativa di due ioni per una resina. Questa dipende dalla carica e dalla solvatazione dei 2 ioni: maggiore la carica e maggiore è la loro affinità. Maggiore è la sfera di solvatazione, minore è l’ affinità per la resina, in quanto si abbassa la capacità di interagire con essa. E` stata stabilita una scala arbitraria di affinità da parte di alcuni cationi dando valore K= 1 al Li+ Li+=1; H+ = 1.2; Na+ 1.98; NH4+ = 2.55 ; K+ = 2.9
![K= [As]*[Br]/[Ar] * [Bs]](http://slideplayer.it/slide/10647384/36/images/29/K%3D+%5BAs%5D%2A%5BBr%5D%2F%5BAr%5D+%2A+%5BBs%5D.jpg "Si stabilirà il seguente equilibrio: Ar + Bs As + Br. Questo equilibrio è regolato dalla legge di azione di massa: K= [As]*[Br]/[Ar] * [Bs] K è il coefficiente di selettività. Esso è una misura dell’ affinità relativa di due ioni per una resina. Questa dipende dalla carica e dalla solvatazione dei 2 ioni: maggiore la carica e maggiore è la loro affinità. Maggiore è la sfera di solvatazione, minore è l’ affinità per la resina, in quanto si abbassa la capacità di interagire con essa. E` stata stabilita una scala arbitraria di affinità da parte di alcuni cationi dando valore K= 1 al Li+ Li+=1; H+ = 1.2; Na+ 1.98; NH4+ = 2.55 ; K+ = 2.9.")
30
Una resina a scambio ionico può essere considerata come un acido (o una base) insolubile che dà luogo, tuttavia ai normali equilibri acido-base, ma con la differenza, rispetto agli equilibri degli acidi (o delle basi solubili) che la parte dell’ acido (o della base) che resta attaccata alla resina non va` in soluzione. Possono essere, pertanto, sostituiti soltanto i contro-ioni.

31
Se ci riferiamo, ad esempio, ad una resina solfonica si ha:
n(Res-SO3-H+) + M+ (Res-SO3)n-M+ + nH+ Analogamente nel caso di una resina a base forte: nRes-N(CH3)3+OH- + A-n Res-N(CH3)3 +A-n + nOH- Se ci riferiamo, ad esempio, alla rimozione dello ione calcio da una soluzione diluita ed acida, mediante resina solfonica in cui è lo ione H+ ad essere in conc. molto più elevata in entrambe le fasi, si ha l’ equilibrio: Ca(a)++ + 2H(R)+ Ca(R)++ + 2H(a)+ Dove gli indici (a) e (R) stanno ad indicare rispettivamente le specie nella fase acquosa o sulla resina. La costante K dell’ equilibrio: K= [Ca++](R) * [H+](a)^2 / [Ca++](a)*[H+]^2 (R) Rappresenta il coefficiente di selettività per la reazione di scambio tra ione calcio e ione idrogeno e fornisce una misura dell’ affinità relativa dei due ioni per la resina.
 + M+ (Res-SO3)n-M+ + nH+ Analogamente nel caso di una resina a base forte: nRes-N(CH3)3+OH- + A-n Res-N(CH3)3 +A-n + nOH- Se ci riferiamo, ad esempio, alla rimozione dello ione calcio da una soluzione diluita ed acida, mediante resina solfonica in cui è lo ione H+ ad essere in conc. molto più elevata in entrambe le fasi, si ha l’ equilibrio: Ca(a)++ + 2H(R)+ Ca(R)++ + 2H(a)+ Dove gli indici (a) e (R) stanno ad indicare rispettivamente le specie nella fase acquosa o sulla resina. La costante K dell’ equilibrio: K= [Ca++](R) * [H+](a)^2 / [Ca++](a)*[H+]^2 (R) Rappresenta il coefficiente di selettività per la reazione di scambio tra ione calcio e ione idrogeno e fornisce una misura dell’ affinità relativa dei due ioni per la resina.")
34
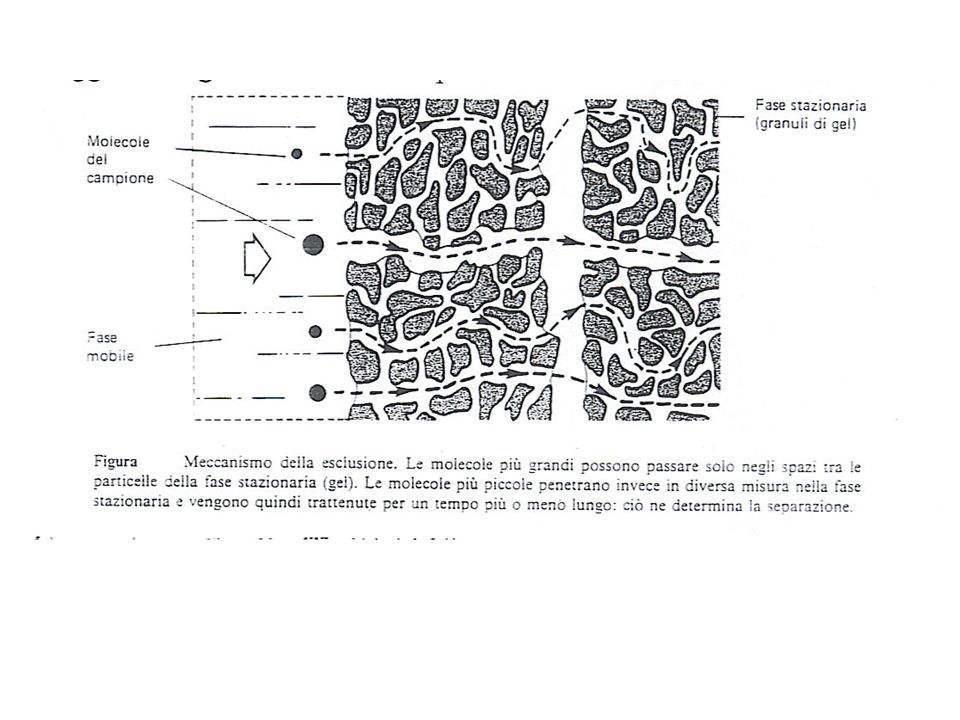
Cromatografia di esclusione ( o gel permeazione).
E’ una tecnica impiegata per la separazione di macromolecole da matrici molto complesse (es liquidi biologici). La fase stazionaria è costituita dal liquido contenuto nelle porosità di un solido polimerico (gel), mentre la fase mobile può essere un solvente organico, acqua, oppure una soluzione tampone. Le molecole di dimensioni inferiori alla luce dei pori delle particelle possono penetrarvi, rimanendo quindi intrappolate dalla fase stazionaria, mentre le rimanenti a peso molecolare maggiore vengono escluse e viaggiano negli interstizi tra le particelle alla velocità dell’ eluente.
. La fase stazionaria è costituita dal liquido contenuto nelle porosità di un solido polimerico (gel), mentre la fase mobile può essere un solvente organico, acqua, oppure una soluzione tampone. Le molecole di dimensioni inferiori alla luce dei pori delle particelle possono penetrarvi, rimanendo quindi intrappolate dalla fase stazionaria, mentre le rimanenti a peso molecolare maggiore vengono escluse e viaggiano negli interstizi tra le particelle alla velocità dell’ eluente.")
36
Cromatografia planare:
Si ratta di tecniche in cui i soluti non escono dal sistema cromatografico, ma vengono messi in evidenza al termine dello sviluppo nella loro posizione finale direttamente sul foglio o sullo strato sottile; sono adatte a scopi analitici o preparativi ( in casi particolari). Cromatografia su carta E’ una cromatografia di ripartizione, in cui il supporto è rappresentato dalla carta, e la fase fissa dall’ acqua trattenuta dalle fibre di cellulosa. Questa tecnica è stata praticamente soppiantata dalla cromatografia su strato sottile. Cromatografia su strato sottile TLC In generale lavora in adsorbimento, con la fase stazionaria solida fissata ad un supporto planare di vetro o plastica o alluminio (lastrina). L’ eluente, salendo per capillarità nella camera di sviluppo, trascina le sostanze che migrano in misura diversa a seconda del grado di ritenzione relativo nel sistema. Le sostanze separate (se incolori) vengono evidenziate con rivelatori universali (H2SO4, KMnO4, I2,, reattivo molibdico) o selettivi (ninidrina, 2-4 dinitrofenilidrazina). Un metodo universale non distruttivo è l’ uso di fluorescenza: se la sostanza non è di per sé fluorescente, si utilizzano lastre contenenti un additivo fluorescente; esaminando le lastre con lampade a UV a 254 nm o 366 nm, si noterà una fluorescenza attenuata in corrispondenza dell’ addensamento delle sostanze.
. Cromatografia su carta. E’ una cromatografia di ripartizione, in cui il supporto è rappresentato dalla carta, e la fase fissa dall’ acqua trattenuta dalle fibre di cellulosa. Questa tecnica è stata praticamente soppiantata dalla cromatografia su strato sottile. Cromatografia su strato sottile TLC. In generale lavora in adsorbimento, con la fase stazionaria solida fissata ad un supporto planare di vetro o plastica o alluminio (lastrina). L’ eluente, salendo per capillarità nella camera di sviluppo, trascina le sostanze che migrano in misura diversa a seconda del grado di ritenzione relativo nel sistema. Le sostanze separate (se incolori) vengono evidenziate con rivelatori universali (H2SO4, KMnO4, I2,, reattivo molibdico) o selettivi (ninidrina, 2-4 dinitrofenilidrazina). Un metodo universale non distruttivo è l’ uso di fluorescenza: se la sostanza non è di per sé fluorescente, si utilizzano lastre contenenti un additivo fluorescente; esaminando le lastre con lampade a UV a 254 nm o 366 nm, si noterà una fluorescenza attenuata in corrispondenza dell’ addensamento delle sostanze.")
37
Il fattore che regola la velocità di migrazione del sistema cromatografico è il coefficiente di ripartizione. In TLC un parametro di più agevole valutazione (correlato a K) è l’ Rf, definito come il rapporto tra la distanza percorsa dal soluto e quella percorsa dal fronte del solvente sulla lastra.
 è l’ Rf, definito come il rapporto tra la distanza percorsa dal soluto e quella percorsa dal fronte del solvente sulla lastra..")
38
HPLC Il nome cromatografia liquida ad elevate prestazioni (HPLC) distingue questa tecnica dai metodi tradizionali di cromatografia liquida su colonna di vetro, ancora in uso a scopi preparativi. L’ HPLC è la tecnica di separazione analitica più usata, in quanto presenta i seguenti vantaggi: Elevata sensibilità Vasta applicabilità (si adatta a prodotti polari, termolabili, non volatili, ad elevato peso molecolare, quindi alla maggior pare dei prodotti di interesse scientifico ed industriale). Accuratezza nelle separazioni quantitative. I principi generali alla base dell’ efficienza cromatografica possono essere applicati all’ HPLC
 distingue questa tecnica dai metodi tradizionali di cromatografia liquida su colonna di vetro, ancora in uso a scopi preparativi. L’ HPLC è la tecnica di separazione analitica più usata, in quanto presenta i seguenti vantaggi: Elevata sensibilità. Vasta applicabilità (si adatta a prodotti polari, termolabili, non volatili, ad elevato peso molecolare, quindi alla maggior pare dei prodotti di interesse scientifico ed industriale). Accuratezza nelle separazioni quantitative. I principi generali alla base dell’ efficienza cromatografica possono essere applicati all’ HPLC.")
39
Strumentazione per HPLC:
Le apparecchiature sono più complesse e costose di quelle usate per altre tecniche, in quanto sono richieste pressioni di lavoro elevate per vincere la resistenza al flusso della fase mobile da parte del micro-impaccamento. La strumentazione richiede quindi un sistema per il pompaggio dell’ eluente a pressioni elevate (fino a 430 atm).
.")
40
I componenti principali sono:
1) Contenitori per la fase mobile e sistemi per il trattamento dei solventi (gorgogliatori, filtri, miscelatori con dispositivi per realizzare i gradienti di eluizione)
 Contenitori per la fase mobile e sistemi per il trattamento dei solventi (gorgogliatori, filtri, miscelatori con dispositivi per realizzare i gradienti di eluizione)")
41
2) Pompa (alternativa a pistone, a siringa (spostamento) o pneumatica)
3)Sistema di introduzione del campione (valvola di iniezione) 4)Colonna di protezione (posta prima della colonna analitica, trattiene il particolato ed i contaminanti contenuti nei solventi) 5)Colonna (lunghezza cm , diametro 4-10 mm; riempimento di microparticelle porose, con dimensioni da 3 a 10 micrometri di silice , allumina o resine scambiatrici) di metallo o vetroborosilicato. L’ approccio in fase normale prevede l’ uso di una fase mobile non polare su di una fase stazionaria polare: l’ uso di queste condizioni comporta un minor numero di parametri da ottimizzare. La cromatografia a fase inversa impiega invece una fase mobile polare (miscele di metanolo, acetonitrile, acqua) su fase stazionaria apolare; è disponibile un gran numero di fasi fisse, la cui scelta consente separazioni molto selettive.
Sistema di introduzione del campione (valvola di iniezione) 4)Colonna di protezione (posta prima della colonna analitica, trattiene il particolato ed i contaminanti contenuti nei solventi) 5)Colonna (lunghezza cm , diametro 4-10 mm; riempimento di microparticelle porose, con dimensioni da 3 a 10 micrometri di silice , allumina o resine scambiatrici) di metallo o vetroborosilicato. L’ approccio in fase normale prevede l’ uso di una fase mobile non polare su di una fase stazionaria polare: l’ uso di queste condizioni comporta un minor numero di parametri da ottimizzare. La cromatografia a fase inversa impiega invece una fase mobile polare (miscele di metanolo, acetonitrile, acqua) su fase stazionaria apolare; è disponibile un gran numero di fasi fisse, la cui scelta consente separazioni molto selettive.")
42
6)Rivelatore (ad assorbimento UV, a fluorescenza, ad indice di rifrazione , elettrochimico).
Ottimizzazione delle condizioni HPLC di ripartizione Spesso in cromatografia di ripartizione si ricorre ad una fase stazionaria di polarità simile a quella dell’ analita, e ad una fase mobile di polarità molto diversa. Questa procedura dà risultati migliori rispetto a quella che impiega la fase mobile a polarità simile a quella del soluto. Nella scelta della fase mobile più idonea, si può utilizzare l’ approccio del fattore singolo, in cui a turno viene variato uno dei seguenti parametri (composizione della fase mobile, pH, forza ionica) o quello multifattoriale. A differenza della gas cromatografia, in LC la natura della fase mobile influenza in modo critico la separazione.
 o quello multifattoriale. A differenza della gas cromatografia, in LC la natura della fase mobile influenza in modo critico la separazione.")
43
Fattori strutturali che governano la velocità di eluizione in HPLC
Eluizione di composti neutri Per una sostanza neutra il tempo di ritenzione dipende dal bilancio fra le caratteristiche lipofile e quelle idrofile; l’ ordine di eluizione dei componenti di una miscela dipende dalla natura della fase stazionaria, la velocità di eluizione è controllata dalla forza della fase mobile. Hplc a fase normale (colonna a fase fissa polare: gel di silice) le sostanze polari vengono trattenute maggiormente rispetto a quelle apolari HPLC a fase inversa le sostanze apolari vengono trattenute maggiormente rispetto a quelle polari.
 le sostanze polari vengono trattenute maggiormente rispetto a quelle apolari. HPLC a fase inversa le sostanze apolari vengono trattenute maggiormente rispetto a quelle polari.")
44
2. Eluizione di composti ionizzabili
Per quelle sostanze il cui grado di ionizzazione dipende dal pH, inclusa quindi la maggioranza dei composti di interesse farmaceutico, la velocità di eluizione è controllata dal pH della fase mobile. L’ instabilità chimica delle fasi fisse per valori estremi di pH ne limita le variazioni nell’ intervallo L’ ottimizzazione della separazione cromatografica tramite controllo del pH trova applicazione soprattutto nell’ hplc in fase inversa. Gli effetti del pH sui tempi di ritenzione non sono sempre spiegabili e prevedibili: molti farmaci infatti in fase inversa vengono trattenuti dalla fase stazionaria anche se completamente ionizzati. Gli effetti maggiori dell’ alterazione del pH sui tempi di ritenzione si hanno per variazioni di 1 unità di pH al di sopra od al di sotto del pKa dell’ analita.

45
HLPC Chirale: La separazione cromatografica di enantiomeri è importante nel controllo di qualità dei farmaci enantiomericamente puri nonchè in studi bioanalitici in cui si debbano esaminare separatamente le farmacocinetiche di composti enantiomerici. La separazione mediante HPLC chirale si basa sulla formazione temporanea di complessi diastereomerici all’ interno della fase stazionaria chirale, caratterizzati da un diverso grado di ritenzione. Non esiste una fase chirale universale, ma sono disponibili in commercio numerosi tipi di fasi stazionarie in grado di separare un gran numero di coppie di enantiomeri. Secondo il modello di interazione basato sui tre punti di contatto, uno solo dei due enantiomeri presenta tutti e tre i sostituenti A, B, e C, nella disposizione spaziale adatta ad interagire con i corrispondenti siti della fase stazionaria, mentre l’ antipodo può interagire con non più di due gruppi con la fase chirale, comunque sia orientato.

46
Le fasi stazionarie chirali più versatili sono le fasi di Pirkle, in cui un residuo di amminoacido è legato con la porzione carbossilica a silice amminopropilica e con l’ amminogruppo all’ a naftil etilammina tramite un ponte ureico. Le interazioni che si stabiliscono tra la fase e ligando sono essenzialmente riconducibili al modello basato sui tre punti di contatto.

47
Gas cromatografia La GC riveste un ruolo importante a scopi analitici qualitativi e, nonostante sia stata sopravanzata in generale come tecnica quantitativa dall’ HPLC, si presta ad alcune analisi quantitative specifiche. I campi di utilizzo sono le scienze ambientali, le industrie alimentari e profumiera, le analisi microbiologiche e cliniche, l’ industria farmaceutica. I principi su cui si basa la GC sono gli stessi trattati precedentemente. Tuttavia in GC, data la comprimibilità dei gas che costituiscono la fase mobile V’r non rappresenta il volume di ritenzione corretto effettivo di un composto; bisogna introdurre un fattore di correzione che tenga conto della caduta di pressione tra l’ ingresso e uscita dalla colonna: Vr netto = V’r * j I meccanismi di separazione che si sfruttano sono l’ adsorbimento, la ripartizione e l’ esclusione molecolare.

48
Vantaggi della GC rispetto alla LC:
Fase mobile gassosa ( a bassa viscosità) Rapido instaurarsi delle condizioni di equilibrio Disponibilità di una vasta gamma di detectors molto efficienti Riutilizzazione della colonna Svantaggi: Applicabilità ai soli composti volatili Fase mobile con sola funzione di trasporto Costi di esercizio (gas).
 Rapido instaurarsi delle condizioni di equilibrio. Disponibilità di una vasta gamma di detectors molto efficienti. Riutilizzazione della colonna. Svantaggi: Applicabilità ai soli composti volatili. Fase mobile con sola funzione di trasporto. Costi di esercizio (gas).")
49
La separazione si basa quindi sulle diverse tensioni di valore e/o le diverse affinità degli analiti per la fase stazionaria (colonne selettive e non). Strumentazione: Un gascromatografo è costituito da: Un sistema di alimentazione del carrier Una camera riscaldata di iniezione / vaporizzazione Una camera termostatata (in modo autonomo rispetto all’ iniettore) contenente la colonna La colonna Il rivelatore Un registratore /integratore Dispositivi vari (regolatori di flusso del carrier, timer per la programmazione della temperatura, ecc
 contenente la colonna. La colonna. Il rivelatore. Un registratore /integratore. Dispositivi vari (regolatori di flusso del carrier, timer per la programmazione della temperatura, ecc.")
50
La camera di vaporizzazione ed il rivelatore sono mantenuti a temperatura più alte rispetto a quella della colonna, che viene fissata a 10-20°C al di sopra del p.e. del composto meno voltatile della miscela da analizzare. Il limite superiore di temperatura dipende dalla natura della fase stazionaria (fenomeno del bleeding). Fase mobile: La fase mobile è costituita dal gas di trasporto (Carrier): idrogeno, azoto, elio, argon, anidride carbonica.
. Fase mobile: La fase mobile è costituita dal gas di trasporto (Carrier): idrogeno, azoto, elio, argon, anidride carbonica.")
51
Requisiti fase mobile:
Inerzia chimica Purezza (assenza di umidità , ossigeno , idrocarburi) Compatibilità con il rivelatore Costo non eccessivo Densità compatibile con il tipo di separazione da effettuare. Fasi stazionarie: Fasi stazionarie per GSC: gel di silice, allumina, carbone attivo, setacci molecolari In genere le fasi sono supportate su materiali inerti, termostabili e finemente suddivisi (silice, celite), opportunamente deattivati e derivatizzati. Esiste in commercio una vasta gamma di fasi fisse (siliconi, poliglicoli, ammidi e nitrili, polieteri , esteri e poliesteri, Idrocarburi , (fasi miste), classificate in base alla polarità.
 Compatibilità con il rivelatore. Costo non eccessivo. Densità compatibile con il tipo di separazione da effettuare. Fasi stazionarie: Fasi stazionarie per GSC: gel di silice, allumina, carbone attivo, setacci molecolari. In genere le fasi sono supportate su materiali inerti, termostabili e finemente suddivisi (silice, celite), opportunamente deattivati e derivatizzati. Esiste in commercio una vasta gamma di fasi fisse (siliconi, poliglicoli, ammidi e nitrili, polieteri , esteri e poliesteri, Idrocarburi , (fasi miste), classificate in base alla polarità.")
53
Colonne capillari Queste colonne ,(in metallo, silice fusa o vetro) sono caratterizzate da diametri interni ridottissimi (da ‘.1 a 0.8 mm) e lunghezze notevoli (fino a 100 m). Si utilizzano ormai di routine nelle analisi di miscele incognite e quando sia richiesta un’ efficienza elevata. Ne esistono di vari tipi con caratteristiche e prestazioni diverse, riconducibili a tre categorie fondamentali: WCOT (wall cated open tubula), in cui la fase stazionaria riveste direttamente la parete SCOT (support coated open tubular) , caratterizzate da un supporto solido (cloruro sodico microcristallino) su cui è fissata la fase fissa PLOT (porous layer open tubular), contenenti uno strato di solido poroso come fase stazionaria (adsorbimento)
 sono caratterizzate da diametri interni ridottissimi (da ‘.1 a 0.8 mm) e lunghezze notevoli (fino a 100 m). Si utilizzano ormai di routine nelle analisi di miscele incognite e quando sia richiesta un’ efficienza elevata. Ne esistono di vari tipi con caratteristiche e prestazioni diverse, riconducibili a tre categorie fondamentali: WCOT (wall cated open tubula), in cui la fase stazionaria riveste direttamente la parete. SCOT (support coated open tubular) , caratterizzate da un supporto solido (cloruro sodico microcristallino) su cui è fissata la fase fissa. PLOT (porous layer open tubular), contenenti uno strato di solido poroso come fase stazionaria (adsorbimento)")
54
Sono generalmente collegate a rivelatori a ionizzazione di fiamma (FID) o a cattura di elettroni (ECD), più efficienti di quelli a termoconducibilità (TCD) Sistemi di iniezione: I campioni liquidi possono essere iniettati direttamente, generalmente previa diluizione all’ 1-10% in un solvente volatile, mentre quelli solidi debbono essere preventivamente solubilizzati: Colonne impaccate Il campione viene introdotto in colonna con una microsiringa (1-10 microL) attraverso un setto di gomma. Colonne capillari: Introduzione in colonna con microsiringa tramite un sistema di iniezione.
 attraverso un setto di gomma. Colonne capillari: Introduzione in colonna con microsiringa tramite un sistema di iniezione.")
55
Rivelatori: La composizione del flusso gassoso in uscita dalla colonna deve essere “Letta” per rilevare i singoli analiti. La lettura viene fatta mediante rivelatori, cioè sistemi che forniscono un segnale quando cambia la composizione del flusso di gas in uscita dalla colonna e forniscono una risposta che è funzione della quantità di sostanza presente nel gas di trasporto. Recentemente sono apparsi al posto dei rivelatori, direttamente degli spettrometri di massa, in grado di effettuare l’ analisi in tempo reale. I rivelatori più usati sono:

56
RIVELATORI UNIVERSALI:
rivelatore a conducibilità termica TCD: questo rivelatore si basa sulla variazione di resistenza prodotta su un sottile filo metallico contenuto in un microcanale mantenuto ad un certo potenziale, dal passaggio della miscela di trasporto di gas/analita, rispetto ad uno analogo percorso solo dal gas di trasporto I due fili metallici sono due dei rami di un ponte di Wheatstone che viene disequilibrato dalla piccola forza elettromotrice prodotta dalla variazione di resistenza e che si riequilibra con una quantità di corrente proporzionale alla quantità di sostanza presente nel gas di trasporto. Ha ridotta sensibilità:

57
Rivelatore a ionizzazione di fiamma FID: è il rivelatore più usato
Rivelatore a ionizzazione di fiamma FID: è il rivelatore più usato. Nel fid l’ effluente in uscita dalla colonna entra mescolato con idrogeno, in un ugello in cui viene bruciato in presenza di aria per produrre una fiamma che si trova tra le espansioni di un campo elettrico, i cui elettrodi sono costituiti dall’ ugello stesso e da una placca posta sopra la fiamma. Quando le molecole dell’ analita entrano nella fiamma, si genera una certa quantità di ioni che producono un incremento di corrente, la quale viene misurato dopo opportuna amplificazione. E’ molto sensibile, sino a 0.1 ng.

58
Registratori: Il segnale analogico proveniente dal rivelatore viene convertito in segnale digitale dal registratore, che trasforma le zone a concentrazione elevata dei componenti separati in picchi cromatografici. I tempi di ritenzione vengono presi come parametri qualitativi (in confronto con degli standard), mentre le aree relative dei picchi si prestano a misure quantitative.
, mentre le aree relative dei picchi si prestano a misure quantitative.")
59
Cromatografia con fluidi supecritici (SFC)
La prerogativa di questo tipo di analisi è l’ impiego di un fluido supercritico come fase mobile. Cos’è un fluido supercritico: Per un dato composto il passaggio di stato liquido-gas e viceversa avviene in un ambito definito di valori di pressione e temperatura. Al di sopra di un certo valore di tamperatura Tc (temperatura critica) una sostanza gassosa non è più ottenibile allo stato liquido, qualunque sia il valore di pressione applicato. I valori di Tc e Pc (pressione critica, corrispondente al valore di pressione richiesta per liquefare il gas alla temperatura Tc) individuano per un dato composto il cosiddetto punto critico C; in queste condizioni gas e liquido presentano la stessa densità. I valori di pressione e temperatura superiori a Tc e Pc definiscono il dominio dello stato di fluido supercritico.
 una sostanza gassosa non è più ottenibile allo stato liquido, qualunque sia il valore di pressione applicato. I valori di Tc e Pc (pressione critica, corrispondente al valore di pressione richiesta per liquefare il gas alla temperatura Tc) individuano per un dato composto il cosiddetto punto critico C; in queste condizioni gas e liquido presentano la stessa densità. I valori di pressione e temperatura superiori a Tc e Pc definiscono il dominio dello stato di fluido supercritico.")
60
Fasi mobili: Come fluidi supercritici si utilizzano comunemente la CO2 (Tc = 31°C, Pc = 7400 KPa), N2O ed NH3. Le proprietà di un fluido supercritico sono intermedie tra quelle di un liquido (solvatazione) e quelle di un gas (viscosità). Un vantaggio di questo tipo di cromatografia è che operando sulla pressione è possibile modificare la densità e quindi il potere solvente della fase mobile. Un gradiente di pressione in SFC equivale ad un gradiente di eluizione in HPLC o ad un gradiente di temperatura in GC. A causa della bassa polarità delle fasi mobili, spesso si aggiunge un composto organico (metanolo, acido formico od acetonitrile) per modificarne le proprietà eluenti.
 e quelle di un gas (viscosità). Un vantaggio di questo tipo di cromatografia è che operando sulla pressione è possibile modificare la densità e quindi il potere solvente della fase mobile. Un gradiente di pressione in SFC equivale ad un gradiente di eluizione in HPLC o ad un gradiente di temperatura in GC. A causa della bassa polarità delle fasi mobili, spesso si aggiunge un composto organico (metanolo, acido formico od acetonitrile) per modificarne le proprietà eluenti.")
61
Vantaggi della SFC: Si presta alla separazione di composti termolabili e/o ad alto peso molecolare E’ una tecnica complementare ad LC e GC , che si usa su un diverso meccanismo di ritardo (dissoluzione – precipitazione) Maggiore rapidità di esecuzione rispetto all’ hplc Svantaggi: L’ efficienza è inferiore a quella ottenibile in GC con colonne capillari Utilizza una strumentazione ibrida GC-HPLC, complessa e costosa L’ impiego di pressioni operative elevate e potenzialmente pericolose.
 Maggiore rapidità di esecuzione rispetto all’ hplc. Svantaggi: L’ efficienza è inferiore a quella ottenibile in GC con colonne capillari. Utilizza una strumentazione ibrida GC-HPLC, complessa e costosa. L’ impiego di pressioni operative elevate e potenzialmente pericolose.")
62
Cenno all’ analisi quantitativa mediante cromatografia:
L’ applicazione principale delle tecniche cromatografiche è l’ analisi quantitativa dei costituenti di una miscela: le quantità sono correlabili all’ altezza od all’ area dei picchi corrispondenti nel cromatogramma. La determinazione della concentrazione di uno o più componenti della miscela può essere effettuata utilizzando vari metodi.

63
Il metodo della standardizzazione esterna per campioni solidi o liquidi molto viscosi. consiste nel preparare soluzioni standard a concentrazione nota, del componente da determinare. Si costruisce una retta o curva di taratura riportando le aree dei picchi di uguali aliquote di varie soluzioni standard in funzione delle concentrazioni corrispondenti degli standard. Si inietta quindi una stessa aliquota di campione, e dalla misura dell’ area si risale alla sua concentrazione attraverso il grafico.

64
Elettroforesi: Le tecniche elettroforetiche sono basate sul movimento di ioni in un campo elettrico. Uno ione di carica q, è sottoposto ad una forza data da F=Eq/d, dove E è il voltaggio (o potenziale elettrico) e d è la distanza tra gli elettrodi. Nel vuoto F causerebbe una accelerazione della molecola. In una soluzione, la molecola è sottoposta al freno della forza di attrito Ff, dovuta al solvente: Ff= 6prhn Dove r è il raggio della molecola carica, h è la viscosità della soluzione, e è la velocità alla quale la molecola carica si sta muovendo. Quindi la velocità di una molecola carica è proporzionale alla carica q e al voltaggio. E, ma è inversamente proporzionale alla viscosità del mezzo e alla distanza tra gli elettrodi. Generalmente l’ elettroforesi non viene condotta in soluzione libera, ma su una matrice di supporto poroso come la poliacrilammide o l’ agarosio, che ritarda i movimenti delle molecole in base alle loro dimensioni in base al diametro dei pori della matrice.
 e d è la distanza tra gli elettrodi. Nel vuoto F causerebbe una accelerazione della molecola. In una soluzione, la molecola è sottoposta al freno della forza di attrito Ff, dovuta al solvente: Ff= 6prhn. Dove r è il raggio della molecola carica, h è la viscosità della soluzione, e. è la velocità alla quale la molecola carica si sta muovendo. Quindi la velocità di una molecola carica è proporzionale alla carica q e al voltaggio. E, ma è inversamente proporzionale alla viscosità del mezzo e alla distanza tra gli elettrodi. Generalmente l’ elettroforesi non viene condotta in soluzione libera, ma su una matrice di supporto poroso come la poliacrilammide o l’ agarosio, che ritarda i movimenti delle molecole in base alle loro dimensioni in base al diametro dei pori della matrice.")
65
Elettroforesi in SDS su gel di poliacrilamide (SDS-Page)
L’ sds è il sodio dodecilsolfato (sodio lauril solfato). La coda idrofobica del dodecilsolfato interagisce fortemente con le catene polipeptidiche. Il numero di SDS legate al polipeptide è proporzionale alla lunghezza (numero di residui amminoacidici) del polipeptide stesso. Ogni dodecilsolfato fornisce due cariche negative. Nel complesso queste cariche coprono qualsiasi carica intrinseca di cui la proteina avrebbe potuto essere dotata.
. La coda idrofobica del dodecilsolfato interagisce fortemente con le catene polipeptidiche. Il numero di SDS legate al polipeptide è proporzionale alla lunghezza (numero di residui amminoacidici) del polipeptide stesso. Ogni dodecilsolfato fornisce due cariche negative. Nel complesso queste cariche coprono qualsiasi carica intrinseca di cui la proteina avrebbe potuto essere dotata.")
66
L’ SDS è anche un detergente che interagisce con il ripiegamento delle proteine (Struttura terziaria). L’ SDS-Page viene di solito svolta in presenza di agenti che riducono i gruppi sulfidrilici, come il betamercaptoetanolo, in modo che i ponti disolfuro tra le catene polipeptidiche vengano rotti. La mobilità elettroforetica delle proteine sottoposte ad SDS-Page è inversamente proporzionale al logaritmo del peso molecolare della proteina. L’ SDS page viene spesso utilizzato per determinare il peso molecolare delle proteine.

67
Focalizzazione al punto isoelettrico:
La focalizzazione isoelettrica è una tecnica elettroforetica atta a separare le proteine in base al loro punto isoelettrico. Una soluzione di anfoliti (elettroliti anfoteri) viene dapprima sottoposta ad elettroforesi su gel, di solito contenuto in un piccolo tubo. La migrazione di queste sostanze in un campo elettrico stabilisce un gradiente di pH lungo il tubo. In seguito viene applicata sul gel una miscela proteica e si riprende la corsa elettroforetica. A mano a mano che le molecole proteiche si muovono lungo il gel, sono sottoposte a un gradiente di pH e migrano fino alla posizione che corrisponde ai loro rispettivi PI. Giunta al suo PI una proteina non possiede più una carica netta, e così non si muove più.
 viene dapprima sottoposta ad elettroforesi su gel, di solito contenuto in un piccolo tubo. La migrazione di queste sostanze in un campo elettrico stabilisce un gradiente di pH lungo il tubo. In seguito viene applicata sul gel una miscela proteica e si riprende la corsa elettroforetica. A mano a mano che le molecole proteiche si muovono lungo il gel, sono sottoposte a un gradiente di pH e migrano fino alla posizione che corrisponde ai loro rispettivi PI. Giunta al suo PI una proteina non possiede più una carica netta, e così non si muove più.")
68
Elettroforesi su gel bidimensionale:
Questa tecnica di separazione di miscele proteiche sfrutta la focalizzazione al punto isoelettrico in una dimensione e l’ SDS-PAGE nella seconda dimensione. Le proteine di una miscela vengono dapprima separate sulla base del PI, per mezzo della focalizzazione al punto isoelettrico eseguita in un tubo di gel di poliacrilamide. Il gel viene quindi rimosso e disteso in cima ad una lastra per SDS-Page, quindi le proteine vengono sottoposte ad elettroforesi SDS gel di poliacrilamimide dove subiscono una separazione sulla base delle loro dimensioni. Il gel può quindi essere colorato, per rivelare la collocazione delle singole proteine.

69
Elettroforesi capillare:
L’ elettroforesi capillare può essere considerata una delle più recenti tecniche separative. L’ elettroforesi, in generale, può essere definita come la migrazione differenziale di specie cariche (ioni ) in soluzione per effetti dell’ applicazione di un campo elettrico; la separazione avviene perchè gli ioni si muovono, sotto l’ azione del campo elettrico applicato, in direzioni diverse (anioni verso il polo positivo e cationi verso il polo negativo)e ad una velocità diversa in base alla loro carica e alla loro massa. Tuttavia se questo processo avviene in soluzione “libera” cioè in un semplice mezzo liquido, non si riesce ad ottenere una separazione degli ioni in bande analitiche, infatti la diffusione termica ed i moti convettivi che ne conseguono, provocano un rapido rimescolamento delle zone in cui gli ioni si separano. Per questo motivo l’ elettroforesi è stata in origine condotta impiegando dei gel (poliacrilammide o agarosio) come mezzi anti-convettivi.
 in soluzione per effetti dell’ applicazione di un campo elettrico; la separazione avviene perchè gli ioni si muovono, sotto l’ azione del campo elettrico applicato, in direzioni diverse (anioni verso il polo positivo e cationi verso il polo negativo)e ad una velocità diversa in base alla loro carica e alla loro massa. Tuttavia se questo processo avviene in soluzione libera cioè in un semplice mezzo liquido, non si riesce ad ottenere una separazione degli ioni in bande analitiche, infatti la diffusione termica ed i moti convettivi che ne conseguono, provocano un rapido rimescolamento delle zone in cui gli ioni si separano. Per questo motivo l’ elettroforesi è stata in origine condotta impiegando dei gel (poliacrilammide o agarosio) come mezzi anti-convettivi.")
70
Questo tipo di tecnica, richiedeva tempi di analisi piuttosto lunghi e ha bassa efficienza di separazione. Infatti l’ applicazione di alte differenze di potenziale non è praticabile poichè lo strato di gel che agisce da suporto anti-convettivo si comporta come una resistenza elettrica; l’ applicazione di una differenza di potenziale genera corrente proporzionale alla sua sezione e per effetto joule si produce una forte quantità di calore. Questo impedisce che possano essere applicati forti campi elettrici; infatti, in tal caso si avrebbe evaporazione del tampone che imbibisce lo strato di gel ed anche il deterioramento delle zone analitiche in cui gli ioni risultano separati.

71
Strumentazione in elettroforesi capillare
Un’ alternativa all’ uso del gel su strato sottile consiste nel realizzare la separazione degli ioni in una soluzione libera contenuta all’ interno di un tubo cilindrico di piccolo diametro (capillare) .Si è osservato infatti che se il tubo ha dimensioni capillari e cioè se il suo diametro interno è dell’ ordine dei micrometri, esso acquista proprietà anticonvettive, e diventa esso stesso in grado di mantenere le zone analitiche separate senza necessità di gel. Ciò permette di ottenere separazioni elettroforetiche in soluzione libera; inoltre, se il diametro del tubo capillare è realmente molto ridotto, è possibile applicare forti differenze di potenziale, senza significativo sviluppo di calore.
 .Si è osservato infatti che se il tubo ha dimensioni capillari e cioè se il suo diametro interno è dell’ ordine dei micrometri, esso acquista proprietà anticonvettive, e diventa esso stesso in grado di mantenere le zone analitiche separate senza necessità di gel. Ciò permette di ottenere separazioni elettroforetiche in soluzione libera; inoltre, se il diametro del tubo capillare è realmente molto ridotto, è possibile applicare forti differenze di potenziale, senza significativo sviluppo di calore.")
72
Figura Il capilare è di solito costituito di materiali quali la silice fusa, di teflon, o vetro pirex, il diametro interno varia da micrometri (la misura più diffusa è di 50) ha una lunghezza variabile, da 20 cm a 1 m. Poichè la migrazione ionica è influenzata dalla temperatura, la strumentazione offre la possibilità di mantenere il capillare (Detto cartuccia) dove, con un sistema a circolazione di aria o di liquido, si può ottenere la termostatazione. b) Compatimenti per il tampone e il generatore Le due estremità del capillare sono introdotte entro due recipienti che contengono entrambi a stessa soluzione tamponata che riempie il capillare. All’ interno di tali recipienti di riserva sono inseriti gli elettrodi del generatore di corrente continua. Il generatore è di tipo molto semplice, a corrente continua e capace di stabilire un potenziale da 0 a 30 KV L’ introduzione di tampone nel capillare è un’ operazione che avviene in modo pneumatico. Mediante una pressione di aria o di azoto. c) Caricamento del campione Per questa operazione, una estremità del capillare viene inseria in un recipiente simile a quelli della riserva di tampone, ma contenente una soluzione del campione in esame. La pressione esercitata sulla testa della soluzione campione deve essere moderata e di breve durata (1-2 secondi) cosi’ da permettere che solo una piccola quantità venga introdotta nel capillare.
 ha una lunghezza variabile, da 20 cm a 1 m. Poichè la migrazione ionica è influenzata dalla temperatura, la strumentazione offre la possibilità di mantenere il capillare (Detto cartuccia) dove, con un sistema a circolazione di aria o di liquido, si può ottenere la termostatazione. b) Compatimenti per il tampone e il generatore. Le due estremità del capillare sono introdotte entro due recipienti che contengono entrambi a stessa soluzione tamponata che riempie il capillare. All’ interno di tali recipienti di riserva sono inseriti gli elettrodi del generatore di corrente continua. Il generatore è di tipo molto semplice, a corrente continua e capace di stabilire un potenziale da 0 a 30 KV. L’ introduzione di tampone nel capillare è un’ operazione che avviene in modo pneumatico. Mediante una pressione di aria o di azoto. c) Caricamento del campione. Per questa operazione, una estremità del capillare viene inseria in un recipiente simile a quelli della riserva di tampone, ma contenente una soluzione del campione in esame. La pressione esercitata sulla testa della soluzione campione deve essere moderata e di breve durata (1-2 secondi) cosi’ da permettere che solo una piccola quantità venga introdotta nel capillare.")
73
Rivelatore Il sitema di rivelazione più diffuso è quello basato sull’ assorbimento spettrofotometrico di radiazioni UV e visibili, in analogia all’ HPLC. La radiazione monocromatica viene fatta attraversare perpendicolarmente alla sezione del capillare in un punto in cui la sua superficie esterna è stata privata della membrana di poliammide. Il rilevatore rivela il passaggio delle varie molecole.

74
Cromatografia di affinità:
Negli ultimi tempi sono state sviluppate strategie di purificazione per affinità delle proteine, che sfruttano la funzione biologica della proteina bersaglio. Nella maggior parte dei casi, le proteine svolgono la loro attività biologica associandosi o formando un complesso con piccole biomolecole specifiche o ligandi, come nel caso di un enzima che si lega al suo substrato. Se questa piccola molecola può essere immobilizzata attraverso un legame covalente ad una matrice insolubile, come un supporto cromatografico quale la cellulosa o la poliacrilammide, allora la proteina di interesse, mostrando affinità per il suo ligando, si legherà e verrà immobilizzata sul sopporto solido stesso. Una volta legata la proteina, le impurezze possono essere lavate via; infine la proteina viene dissociata od eluita dalla matrice attraverso l’ aggiunta di una alta concentrazione di ligando libero alla soluzione.









>")





1 Soluzioni e sospensioni.>")
 Isolamento e Purificazione dei Composti Organici>")


 di cui quella presente in quantità maggiore è definita solvente,>")


