Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Dipartimento di Genetica
Università di Torino Dipartimento di Genetica Biologia e Biochimica barbara pasini Predisposizioni ereditarie allo sviluppo di tumori neuroendocrini MEN2 e MEN1

2
HPT-JT MEN2 CDK-Inhibitors MEN1 VHL carcinoma midollare della tiroide
iperplasia / adenoma paratiroidi MEN1 AIP PRKAR1A SDHB-SDHD SDHC, SDHAF2SDH5, SDHA TMEM127 e NF1 paraganglioma feocromocitoma tumori endocrini pancreatici angiomatosi retinica emangioblastoma SNC carcinoma renale (CC) adenoma ipofisi gastrinomi duodenali carcinoidi ecc. VHL
 adenoma ipofisi. gastrinomi duodenali. carcinoidi. ecc. VHL.")
3
caratteristiche generali delle neoplasie endocrine multiple
autosomiche dominanti combinazione di “lesioni” specifiche istologia / clinica caratteristica PT: iperplasia in MEN2A / adenomi in MEN1 / adenoma cistico HPT-JT PiA: frequente PRL in MEN1 – frequente GH in AIP PNET: spesso secernenti in MEN1 / non secernenti VHL MTC con CCH in MEN2 età caratteristiche di insorgenza più manifestazioni presenti contemporaneamente sviluppo di manifestazioni in successione manifestazioni cutanee, cistiche ecc. malformazioni, dismorfismi (MEN2B) penetranza alta ma espressività variabile correlazione genotipo-fenotipo - MEN2, VHL, SDH (no MEN1)
 penetranza alta ma espressività variabile. correlazione genotipo-fenotipo. - MEN2, VHL, SDH (no MEN1)")
4
neoplasie endocrine multiple
test genetico nelle neoplasie endocrine multiple indicazione al test sospetto clinico: almeno due segni di malattia o famigliarità vantaggio del test genetico rispetto alla diagnosi clinica - paziente giovane alla prima manifestazione - le altre manifestazioni attese sono più tardive - lo screening clinico è costoso / impegnativo - il test genetico è informativo utilità del test confermare la diagnosi (può venire negativo !) correlazioni genotipo-fenotipo stimare il rischio per ulteriori manifestazioni di malattia curare in modo opportuno le manifestazioni di malattia screening dei famigliari a rischio (età precoce) diagnosi prenatale
 correlazioni genotipo-fenotipo. stimare il rischio per ulteriori manifestazioni di malattia. curare in modo opportuno le manifestazioni di malattia. screening dei famigliari a rischio (età precoce) diagnosi prenatale.")
5
MEN 2 (AD) MEN 2A: 95% carcinoma midollare della tiroide
50% feocromocitoma 15-30% iperparatiroidismo (iperplasia >> adenoma) raramente lichen amiloidosico interscapolare malattia di Hirschsprung nervi corneali prominenti
 raramente lichen amiloidosico interscapolare. malattia di Hirschsprung. nervi corneali prominenti.")
6
MEN 2 (AD) MEN 2A - 1 : MTC + pheo + HPT MEN 2A - 2 : MTC + pheo
MEN 2A - 3 : MTC + HPT FMTC : almeno 4-8 casi di MTC in famiglia parenti affetti viventi con sorveglianza clinica negativa per pheo e HPT MEN 2B : insorgenza precoce di MTC con o senza pheo dismorfismi facciali, neuromi delle mucose (labbra) habitus marfanoide, cifoscoliosi ganglioneuromatosi intestinale pectus excavatum, pes cavus
 habitus marfanoide, cifoscoliosi. ganglioneuromatosi intestinale. pectus excavatum, pes cavus.")
7
MEN 2B Spinelli e Bernini
in Endocrinopatie pediatriche di interesse chirurgico R. Domini et al. Andrioli M.

8
proto-oncogene RET (10q11.2)
RTK (receptor tyrosine kinase) Ligandi : fattori neurotrofici della famiglia del transforming growth factor b : GDNF, Neurturina, Persefina, Artemina cadherine-like cys-rich region trans-membrane tyrosine kinase domain 3 isoforms: (ex 19) (ex 20) (ex 21) Co-recettori : Proteine legate al glicosil-fosfatidil-inositolo GFRa1, GFRa2, GFRa3, GFRa4
 Ligandi : fattori neurotrofici della famiglia del. transforming growth factor b : GDNF, Neurturina, Persefina, Artemina. cadherine-like. cys-rich region. trans-membrane. tyrosine kinase. domain. 3 isoforms: (ex 19) (ex 20) (ex 21) Co-recettori : Proteine legate al glicosil-fosfatidil-inositolo. GFRa1, GFRa2, GFRa3, GFRa4.")
9
Mutazioni “germinali” in recettori TK
RET : neoplasie endocrine multiple tipo 2 MET : carcinoma renale ereditario papillare KIT : GIST (gastro-intestinal stromal tumors) famigliari PDGFRa : GIST e neurofibromatosi intestinale “Caratteristiche comuni” : - predisposizioni ai tumori AD - pattern limitato di espressione dei geni - origine dei tumori dalle cellule che li esprimono - possible presenza di difetti di sviluppo
 famigliari. PDGFRa : GIST e neurofibromatosi intestinale. Caratteristiche comuni : - predisposizioni ai tumori AD. - pattern limitato di espressione dei geni. - origine dei tumori dalle cellule che li esprimono. - possible presenza di difetti di sviluppo.")
10
Polimorfismi Mutazioni Cys609 , Cys611 (mut Cys611 + Arg886Trp in cis)
ex 5 Gly321Arg (1 family) Gly533Cys (1 family) Cys855Tyr (HD, MEN2 ??) Lys603 Gln (1 family FMTC – PTC ??) Cys609 , Cys611 (mut Cys611 + Arg886Trp in cis) Cys618, Cys620 Cys630 Asp631Tyr ?? Cys634 (mut Cys634 + Asp631Glu, Arg635Gly, Ala640Gly, Ala641Ser in cis) (mut Cys634 + Val 648Ile in trans in ACTH producing Pheo) Ser649Leu (CCH ??), Lys666Glu (??) Glu768Asp (GAT, GAC) Asn777Ser Val778Ile (multifocal MTC ??), Gln781Arg (FMTC ??) Leu790Phe (TTT, TTC) Val804Leu, Met Val804Met + Tyr806Cys in cis, Val804Met + Ser904Cys in cis Val804Met + Arg844Leu in cis Met848Thr (MTC ??), Ile852Met (MTC ??) Ala883Phe, Ala883Thr (MTC ??) Ser891Ala, Ser904Phe (MTC ??) Arg912Pro (1 family) Met918Thr, Ser922Pro/Tyr (MTC ??) Polimorfismi Mutazioni ex 8 ex 9 ex 10 Glu616del ?? Asp623Lys ?? Asp631Val+His665Gln ?? Ile647 (ATC>ATT) Gly691Ser ex 11 ex 13 Leu769 (CTT>CTG) Tyr791Phe (TAT>TTT) ex 14 Tyr806Cys ?? Ser836 (AGC>AGT) ex 15 Ser904 (TCC>TCG) ex 16
 Gly533Cys (1 family) Cys855Tyr (HD, MEN2 ) Lys603 Gln (1 family FMTC – PTC ) Cys609 , Cys611 (mut Cys611 + Arg886Trp in cis) Cys618, Cys620. Cys630. Asp631Tyr Cys634 (mut Cys634 + Asp631Glu, Arg635Gly, Ala640Gly, Ala641Ser in cis) (mut Cys634 + Val 648Ile in trans in ACTH producing Pheo) Ser649Leu (CCH ), Lys666Glu ( ) Glu768Asp (GAT, GAC) Asn777Ser. Val778Ile (multifocal MTC ), Gln781Arg (FMTC ) Leu790Phe (TTT, TTC) Val804Leu, Met. Val804Met + Tyr806Cys in cis, Val804Met + Ser904Cys in cis. Val804Met + Arg844Leu in cis. Met848Thr (MTC ), Ile852Met (MTC ) Ala883Phe, Ala883Thr (MTC ) Ser891Ala, Ser904Phe (MTC ) Arg912Pro (1 family) Met918Thr, Ser922Pro/Tyr (MTC ) Polimorfismi. Mutazioni. ex 8. ex 9. ex 10. Glu616del Asp623Lys Asp631Val+His665Gln Ile647 (ATC>ATT) Gly691Ser. ex 11. ex 13. Leu769 (CTT>CTG) Tyr791Phe (TAT>TTT) ex 14. Tyr806Cys Ser836 (AGC>AGT) ex 15. Ser904 (TCC>TCG) ex 16.")
11
Mutazioni RET extra-cellulari: livello di rischio 2, 3
Cys609 # Cys611 Cys618 # Cys620 # 12 bp dup ( ) Cys630 Cys634 MEN2A-1: 8% MEN2A-2: 31% MEN2A-3: 20% FMTC : 60 % HSCR: 65 casi ex 10 ex 11 cys-rich region MEN2A-1: 92% MEN2A-2: 69% MEN2A-3: 80% FMTC : 30 % trans-membrane tyrosine kinase domain Effetto biologico : dimerizzazione ligando-indipendente del recettore RET
 Cys630. Cys634. MEN2A-1: 8% MEN2A-2: 31% MEN2A-3: 20% FMTC : 60 % HSCR: 65 casi. ex 10. ex 11. cys-rich region. MEN2A-1: 92% MEN2A-2: 69% MEN2A-3: 80% FMTC : 30 % trans-membrane. tyrosine kinase. domain. Effetto biologico : dimerizzazione ligando-indipendente. del recettore RET.")
12
RET esone 11 p.Cys634Tyr mut wt

13
RET esone 11 p.Cys634Tyr + wt

14
RET esone 11 p.Cys634Tyr

15
RET esone 14 p.Val804Met CT al test genetico basale : 2.60 pg/ml
wt mut CT al test genetico basale : 2.60 pg/ml picco : pg/ml CT al test genetico basale : pg/ml picco : pg/ml

16
RET esone 10 p.Cys618Arg wt wt wt wt wt wt wt wt wt wt

17
wt

18
Mutazioni RET in Cys esone 10 : MEN2A-FMTC e malattia di Hirschsprung
23 famiglie 65 casi

19
aganglionosi del colon distale ostruzione e megacolon a monte
MEN2A – HD aganglionosi del colon distale ostruzione e megacolon a monte MEN2B – ganglioneuromatosi perdita del “tono” muscolare distensione, dilatazione segmentaria megacolon

20
Mutazioni RET intra-cellulari:
Livello di rischio 1 (MEN2B) Effetto biologico: cambiamento della specificità di substrato Livello di rischio 3 (FMTC) (raramente MEN2A) trans- membrane Glu768 Asp Leu790 Phe Tyr791 Phe (variante genetica con modesto effetto biologico) Val804 Leu Val804 Met Ala883 Phe Ser891 Ala Met918 Thr ATP binding domain ex 13 inter-TK ex 14 ex 15 Catalytic core pocket ex 16
 Effetto biologico: cambiamento della. specificità di substrato. Livello di rischio 3 (FMTC) (raramente MEN2A) trans- membrane. Glu768 Asp. Leu790 Phe. Tyr791 Phe (variante genetica con modesto effetto biologico) Val804 Leu. Val804 Met. Ala883 Phe. Ser891 Ala. Met918 Thr. ATP. binding. domain. ex 13. inter-TK. ex 14. ex 15. Catalytic. core. pocket. ex 16.")
21
RET esone 16 p.Met918Thr Nato nel 1998:
- labbra e neurofibromi linguali tipici - dal 2001, accertamenti per ripetuti episodi di stipsi 2007: biopsia lesione lingua e dosaggio calcitonina (187.6 pg/ml vn < 10) cromogranina A (143.7 ng/ml vn < 98) normali valori del paratormone (27.4), calcemia (9.7) e catecolamine urinarie eco tiroide: nodulo di 5 mm del lobo destro e nodulo solido, ipoecogeno, a margini irregolari di 6 x 5 mm del lobo sinistro test da stimolo: calcitoninemia basale di 90.6 ng/l picco al 3' di e al 5' di 811.3, cromo A: 90 ng/ml EI: carcinoma midollare della tiroide bilaterale (dx: 4 mm, sin: 5 mm) associato ad iperplasia delle cellule C, iperplasia reattiva in 12 linfonodi analizzati 2008: calcitonina basale 13 pg/ml (picco a 3’: 58, a 5’: 51), CromoA 117 ng/ml 2009: calcitonina 21 pg/mL, cromoA 91 ng/m
 cromogranina A (143.7 ng/ml vn < 98) normali valori del paratormone (27.4), calcemia (9.7) e catecolamine urinarie. eco tiroide: nodulo di 5 mm del lobo destro e nodulo. solido, ipoecogeno, a margini irregolari di 6 x 5 mm. del lobo sinistro. test da stimolo: calcitoninemia basale di 90.6 ng/l. picco al 3 di e al 5 di 811.3, cromo A: 90 ng/ml. EI: carcinoma midollare della tiroide bilaterale. (dx: 4 mm, sin: 5 mm) associato ad iperplasia delle. cellule C, iperplasia reattiva in 12 linfonodi analizzati. 2008: calcitonina basale 13 pg/ml. (picco a 3’: 58, a 5’: 51), CromoA 117 ng/ml. 2009: calcitonina 21 pg/mL, cromoA 91 ng/m.")
22
RET esone 10 c.1852T>A (p.Cys618Ser) RR 50% MEN2 10% stenosi piloro
< 2% HSCR Nato nel 1968 con stenosi del piloro (32): tumefazione del testicolo sinistro Ecografia: lesione nodulare solida di circa 1,5 cm CEA 71.1 ng/ml (vn < 5), aFP 1.7 UI/ml (vn < 5) BHCG < 1 mUI/ml intervento di orchi-funicolectomia sinistra EI: neoplasia stromale del testicolo (basso grado di atipie, CEA negativo) TC addome completo nella norma CEA 77.3 ng/ml (vn < 5) 2005 (36): CEA 92.6 esame PET: area di captazione in regione paratracheale sinistra alla base del collo cui segue esame ecografico nodulo tiroideo sinistro di 2,9 cm risultato negativo per CTM calcitonina 621 pg/ml, PTH (28) e catecolamine urinarie neg. tiroidectomia totale con linfoadenectomia laterocervicale bilaterale asportazione del timo reimpianto di tessuto paratiroideo EI: carcinoma midollare della tiroide monolocale di 3,5 x 1,8 cm non segnalata iperplasia delle cellule C, pT2N0
: tumefazione del testicolo sinistro. Ecografia: lesione nodulare solida di circa 1,5 cm. CEA 71.1 ng/ml (vn < 5), aFP 1.7 UI/ml (vn < 5) BHCG < 1 mUI/ml. intervento di orchi-funicolectomia sinistra. EI: neoplasia stromale del testicolo. (basso grado di atipie, CEA negativo) TC addome completo nella norma. CEA 77.3 ng/ml (vn < 5) 2005 (36): CEA esame PET: area di captazione in regione paratracheale. sinistra alla base del collo cui segue esame ecografico. nodulo tiroideo sinistro di 2,9 cm risultato negativo per CTM. calcitonina 621 pg/ml, PTH (28) e catecolamine urinarie neg tiroidectomia totale con linfoadenectomia. laterocervicale bilaterale. asportazione del timo. reimpianto di tessuto paratiroideo. EI: carcinoma midollare della tiroide. monolocale di 3,5 x 1,8 cm. non segnalata iperplasia delle cellule C, pT2N0.")
23
RET esone 13 p.Glu798Asp Breast Cancer 58 mut wt

24
mut RET esone 13 p.Leu790Phe Calcitoninemia 9.7 pg/ml

25
RET esone 13 p.Leu790Phe

26
carcinoma midollare della tiroide - apparentemente sporadico
analisi RET nel carcinoma midollare della tiroide - apparentemente sporadico e nel feocromocitoma 138 casi analizzati di CMT sporadico: mutazioni RET in 12 casi (8.7%): 7 in cisteine esone 10 (Cys611, 618, 620) 1 esone 13 (p.Glu768Asp) 3 esone 14 (p.Val804Met) 1 esone 16 (p.Met918Thr metastatico a 29 aa) età alla diagnosi dai 28 ai 77 anni 35 casi analizzati di PHEO sporadici, bilaterali, famigliari: nessuna mutazioni RET (0%) p.s. 8 mutazioni VHL (23%) gene RET 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
: 7 in cisteine esone 10 (Cys611, 618, 620) 1 esone 13 (p.Glu768Asp) 3 esone 14 (p.Val804Met) 1 esone 16 (p.Met918Thr metastatico a 29 aa) età alla diagnosi dai 28 ai 77 anni. 35 casi analizzati di PHEO. sporadici, bilaterali, famigliari: nessuna mutazioni RET (0%) p.s. 8 mutazioni VHL (23%) gene RET")
27
Mutazioni germinali in feocromocitomi (surrene) - paragangliomi
 - paragangliomi")
28
Mutazioni germinali in feocromocitomi (surrene) – paragangliomi maligni
 – paragangliomi maligni")
29
Mutazioni germinali in feocromocitomi (surrene) – paragangliomi “sospetti” ereditari
 – paragangliomi sospetti ereditari")
30
in sospetta MEN2 e altri fenotipi
analisi RET in sospetta MEN2 e altri fenotipi nessuna mutazioni RET in: - 6 casi con CCH - 4 casi di iperparatiroidismo - 3 casi con diade/ triade di Carney CMT famigliare: 1 mut / 3 (33%): esone 14 (p.Val804Met) sospetta MEN2A: 4 mut / 9 (44%): esone 10 (1) e 11 (3) casi negativi: CMT + HPT, PHEO con CCH sospetta MEN2B: 3 mut / 3 (100%): esone 16 (p.Met918Thr) Screening dei famigliari per mutazione RET nota in fam. (73): 26 mutati 47 non portatori gene RET 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
: esone 14 (p.Val804Met) sospetta MEN2A: 4 mut / 9 (44%): esone 10 (1) e 11 (3) casi negativi: CMT + HPT, PHEO con CCH. sospetta MEN2B: 3 mut / 3 (100%): esone 16 (p.Met918Thr) Screening dei famigliari per. mutazione RET nota in fam. (73): 26 mutati. 47 non portatori. gene RET")
31
MEN2: Gestione clinica 611, 618, 620, 634 609, 768, 790, 804, 891

32
Neoplasie endocrine multiple tipo 1 (AD)
Autosomica dominante alta penetranza ma espressività variabile Altre manifestazioni lipomi angiofibromi facciali collagenomi ependimoma feocromocitoma noduli corteccia surrenalica

33
Neoplasie endocrine multiple tipo 1 (AD)
Penetranza del 50% a 20 anni – 95% a 40 anni

34
<> VIPoma: 2% dei casi <> Glucagonoma: 2% dei casi
Gastrinomi: in circa il 40% dei casi sintomi di ZES di solito < 40 anni spesso multipli e < 1 cm triangolo del gastrinoma: duodeno testa del pancreas legamento epatoduodenale (NO gastrici !!!) trattamento alla diagnosi (segni biochimici o > 1 cm) metastasi alla diagnosi nel 50% (pancreas > duodeno) mortalità nel 30% dei casi metastatici al fegato <> Insulinomi: in circa il 10% dei casi tumori per lo più unici e solitamente benigni iper-insulinismo se 1- 4 cm associati a macro-adenomi <> VIPoma: 2% dei casi <> Glucagonoma: 2% dei casi <> Non funzionanti: frequenti
 trattamento alla diagnosi (segni biochimici o > 1 cm) metastasi alla diagnosi nel 50% (pancreas > duodeno) mortalità nel 30% dei casi metastatici al fegato. <> Insulinomi: in circa il 10% dei casi. tumori per lo più unici e solitamente benigni. iper-insulinismo se 1- 4 cm. associati a macro-adenomi. <> VIPoma: 2% dei casi. <> Glucagonoma: 2% dei casi. <> Non funzionanti: frequenti.")
35
gene MEN1 clonato nel 1997 10 esoni in 9 Kb localizzato nel 1988
in 11q13 proteina menina di 610 aa – 67 kDa espressa in molti tessuti localizzazione nucleare nelle cellule quiescenti localizzazione citoplasmatica nelle cellule in divisione

36
Mutazioni germinali MEN1
riportate 1133 mutazioni (459 differenti) 73% mutazioni troncanti 41% delezioni / inserzioni (0/11) 23% nonsenso (1/11) 9% difetti di splicing (6/11) 6% inserzioni / delezioni in frame 20% missenso (4/11) 68% in aa conservati nei geni omologhi 27% in aa non identici ma affini 5% in aa non conservati 1% delezioni genomiche (0/11) assenza di correlazioni tra genotipo e fenotipo (nel FIHP le mutazioni missenso sono il 38%) 4 hot-spots mutazionali (12.3% delle mutazioni)
 73% mutazioni troncanti. 41% delezioni / inserzioni (0/11) 23% nonsenso (1/11) 9% difetti di splicing (6/11) 6% inserzioni / delezioni in frame. 20% missenso (4/11) 68% in aa conservati nei geni omologhi. 27% in aa non identici ma affini. 5% in aa non conservati. 1% delezioni genomiche (0/11) assenza di correlazioni tra genotipo e fenotipo. (nel FIHP le mutazioni missenso sono il 38%) 4 hot-spots mutazionali (12.3% delle mutazioni)")
37
Mutazioni germinali MEN1
77 % dei casi famigliari MEN1 64 % dei casi isolati di MEN1 20 % dei casi famigliari di HPT assenti nei casi famigliari di prolattinoma o acromegalia (gene AIP in 11q13) rare nei casi sporadici di : - gastrinoma (5%) - carcinoide timico (7%) - adenoma delle paratiroidi (< 1%) - prolattinoma (< 1%) angiofibroma (< 1%) 11/71 (15.5%)
 rare nei casi sporadici di : - gastrinoma (5%) - carcinoide timico (7%) - adenoma delle paratiroidi (< 1%) - prolattinoma (< 1%) angiofibroma (< 1%) 11/71 (15.5%)")
38
MEN1 p.Lys557X MEN1 IVS3+1G>A Carcinoide mediastino 36
Tumore endocrino Pancreas 39

39
MEN1 IVS6-2A>G MEN1 p.Arg415X

40
MEN1 IVS4-9G>A

41
?
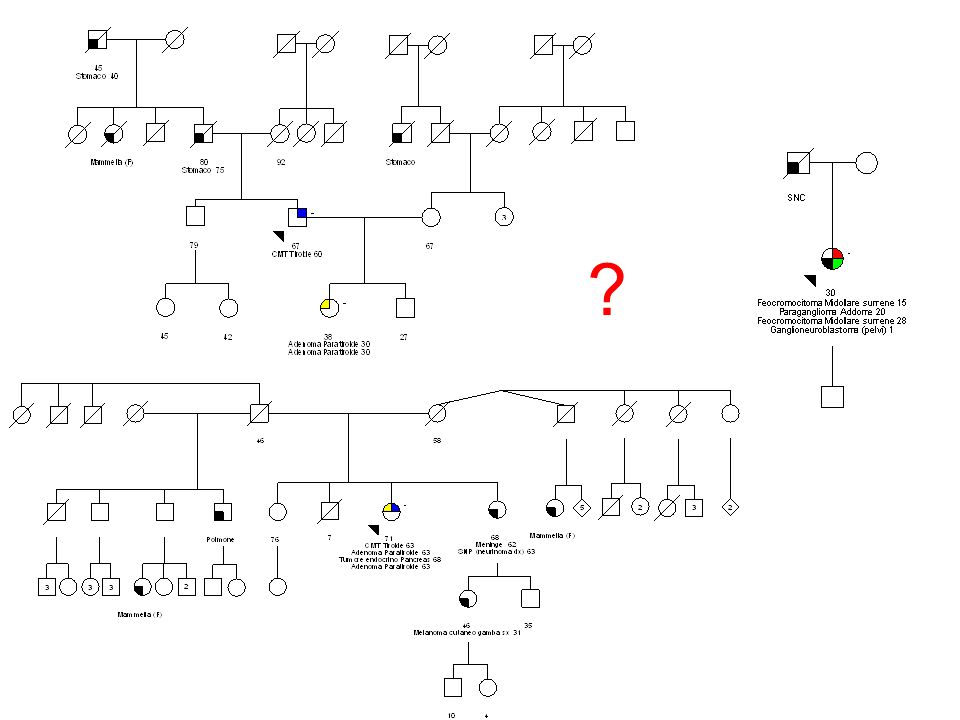
42
MEN1 – like MEN4

Presentazioni simili














>")

>")


