Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Fisiopatologia dell’emostasi: la cascata e dintorni
Prof. Enrico Maria Pogliani 16/4/2014

3
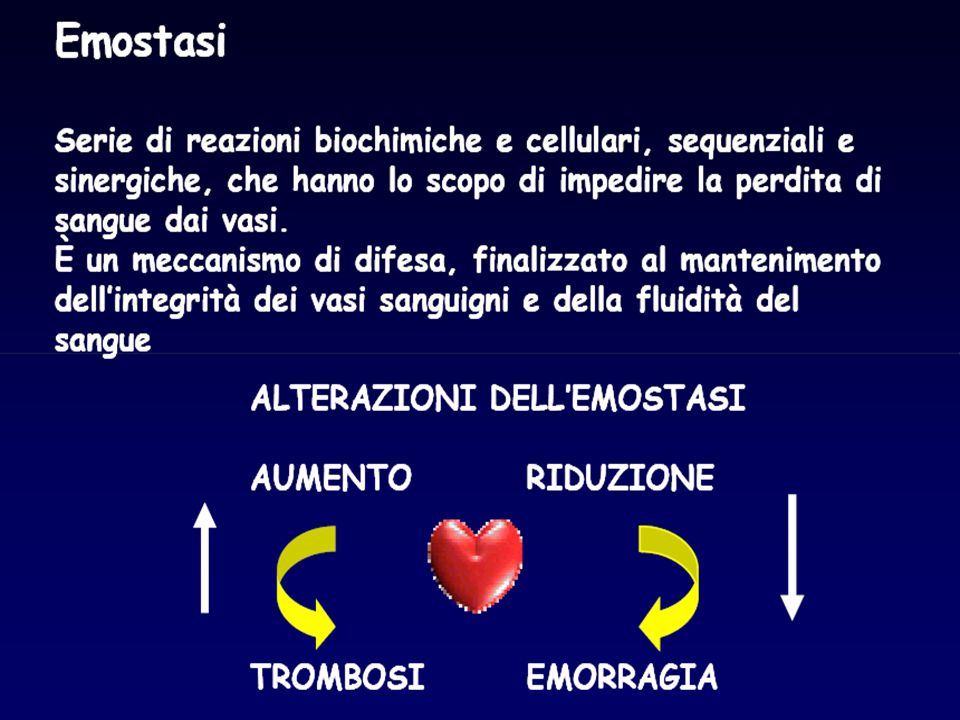
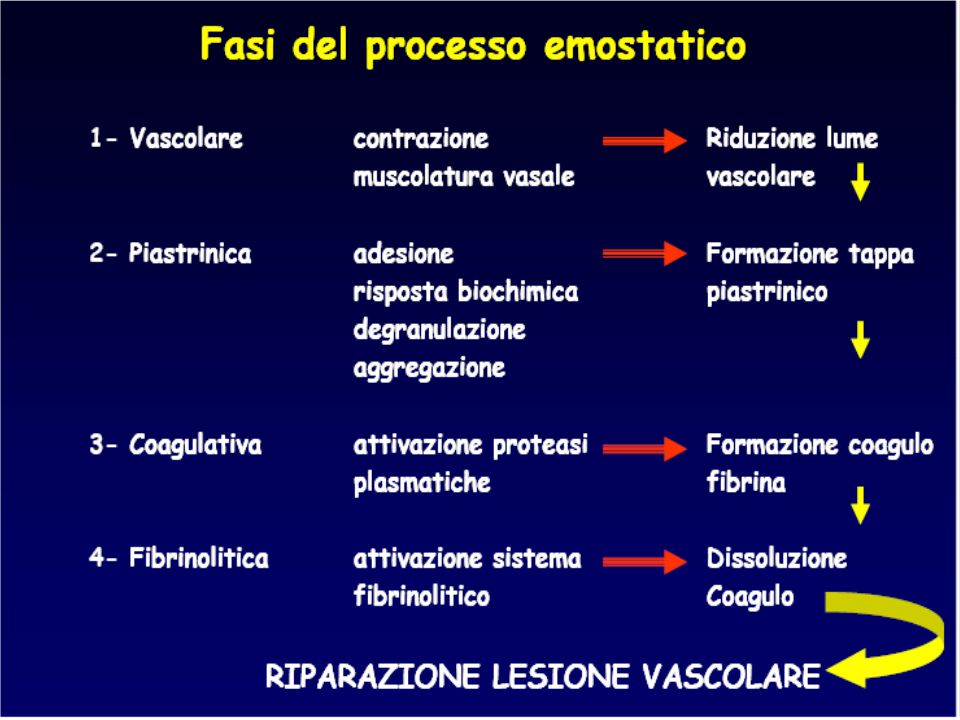
Perché l’emostasi “s’ha da fare”?
…perché è nel programma di studio ..per capirci qualcosa, quando il paziente sanguina …per saper interpretare gli esami di laboratorio perché i disordini congeniti e acquisiti dell’emostasi non sono così rari!! per l’ampia diffusione di farmaci antiaggreganti e anticoagulanti ->alterazioni acquisite dell’emostasi

4
Un po’ di fisiologia….per cominciare

7
Funzione dell’endotelio vascolare
Endothelium-derived relaxing factor

11
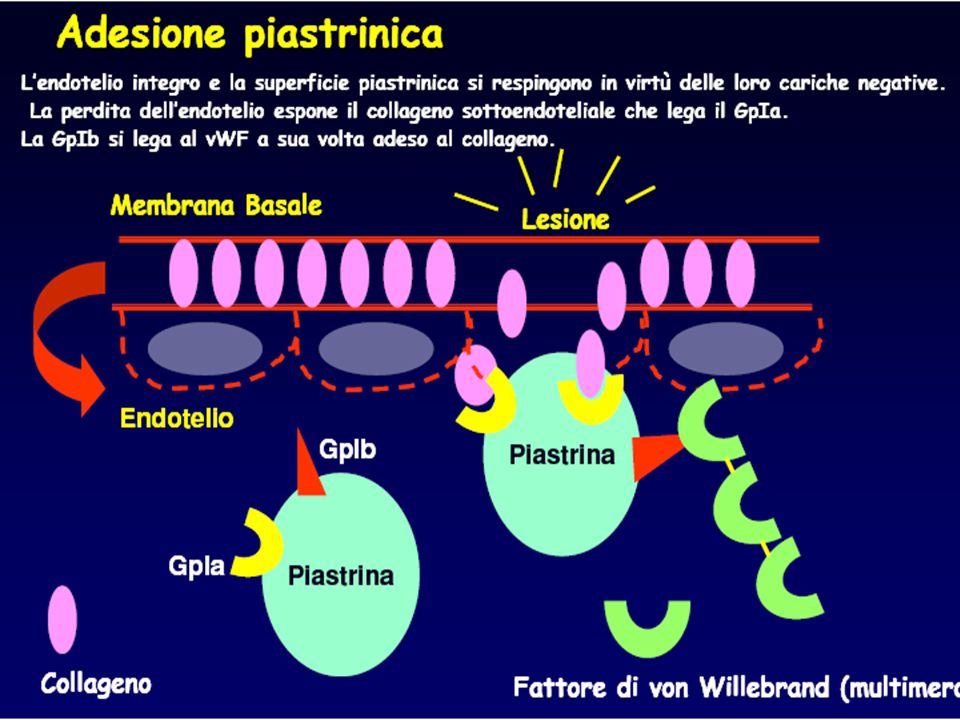
Organelli piastrinici
A atmosfera piastrinica B membrana trilaminare C sistema canalicolare aperto D citoscheletro (sistema microtubulare contrattile ) E sistema tubulare denso F glicogeno G mitocondrio H granuli α ( b-TG , FP4, fibrinogeno, VIII-Ag ) I granuli δ ( ADP, ATP, serotonina, Ca++ ) L granuli λ ( idrolisi, proteasi, catepsine )
 E sistema tubulare denso F glicogeno G mitocondrio H granuli α ( b-TG , FP4, fibrinogeno, VIII-Ag ) I granuli δ ( ADP, ATP, serotonina, Ca++ ) L granuli λ ( idrolisi, proteasi, catepsine )")
17
3

18
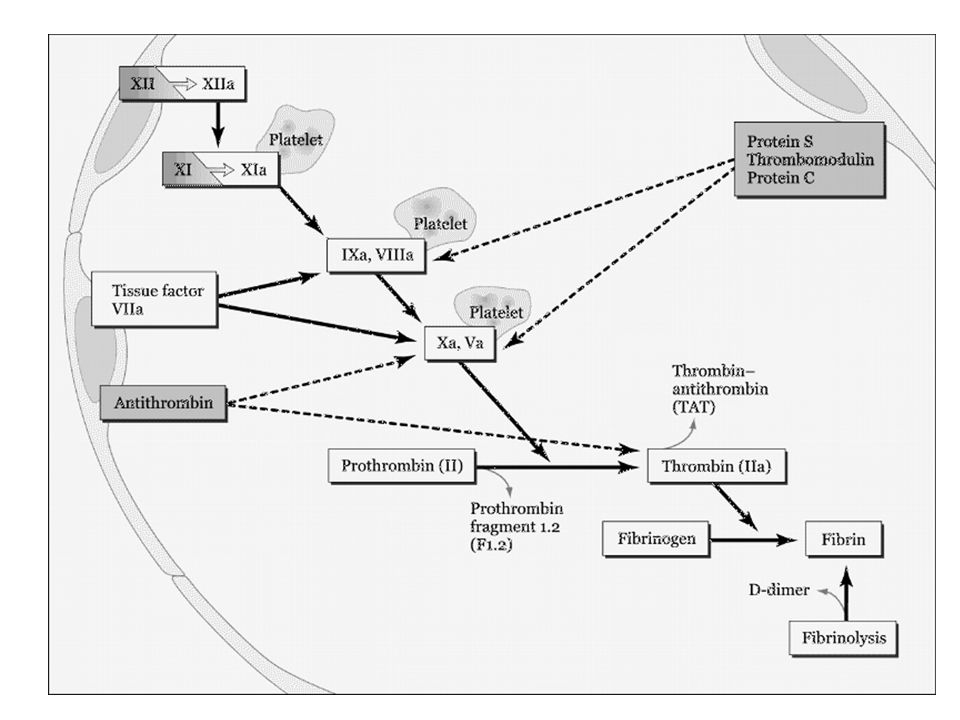
La cascata “moderna” Protrombina

19
Contact Activation (Intrinsic) pathway
Tissue Factor (Extrinsic) pathway Common pathway
 pathway. Common pathway.")
22
4

25
Come si valuta l’emostasi

30
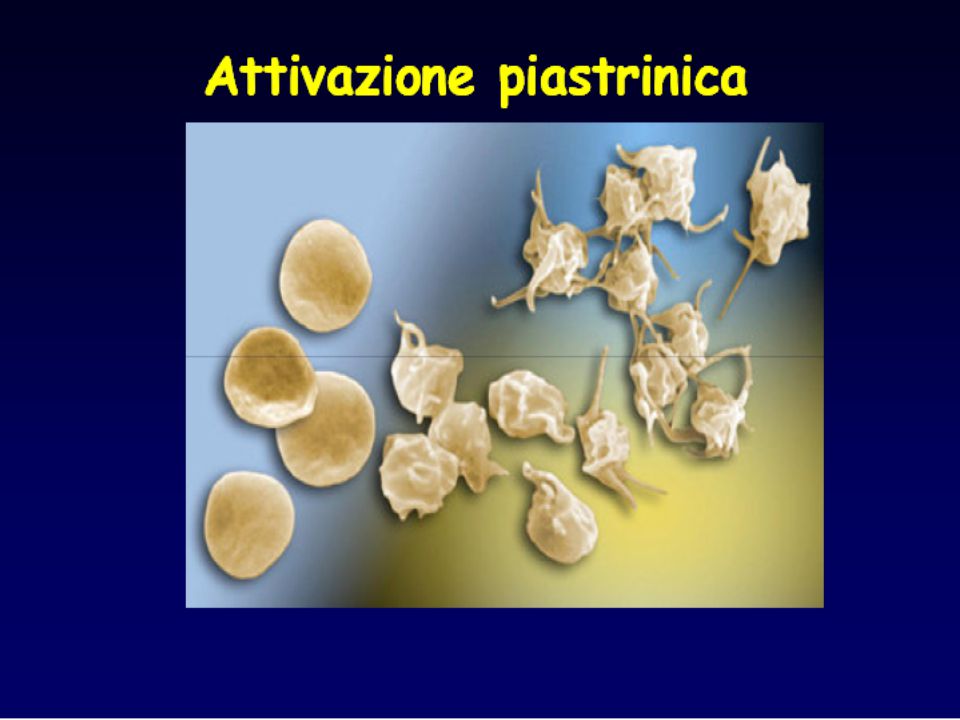
Agregazione piastrinica indotta da ADP
Agregazione piastrinica indotta da trombina

32
Il coagulometro

33
La provetta…

34
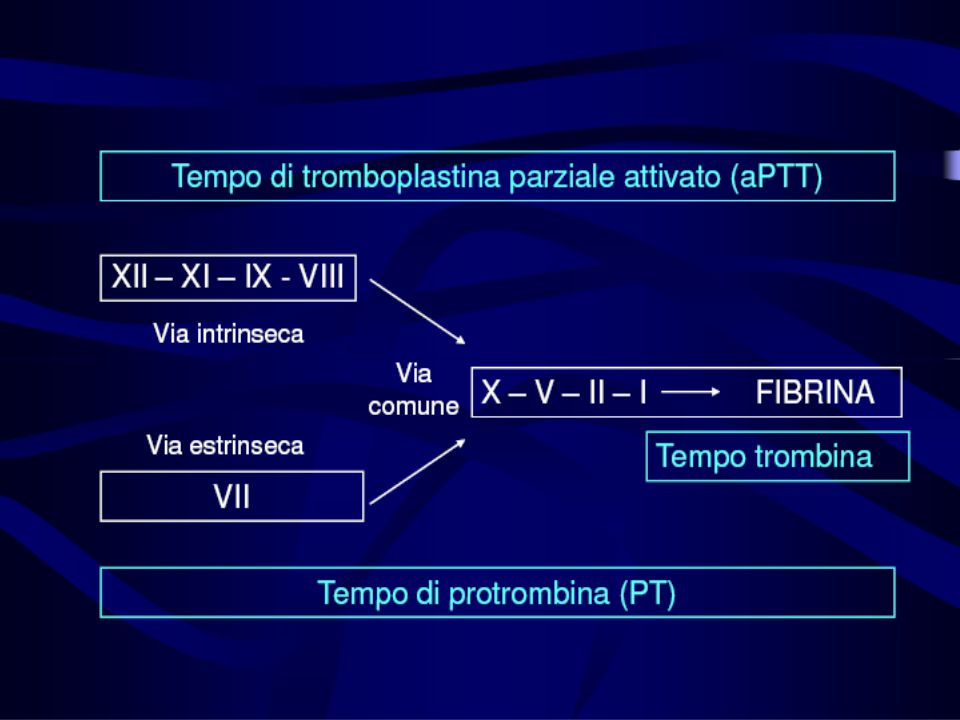
Tempo di tromboplastina parziale attivata (aPTT)
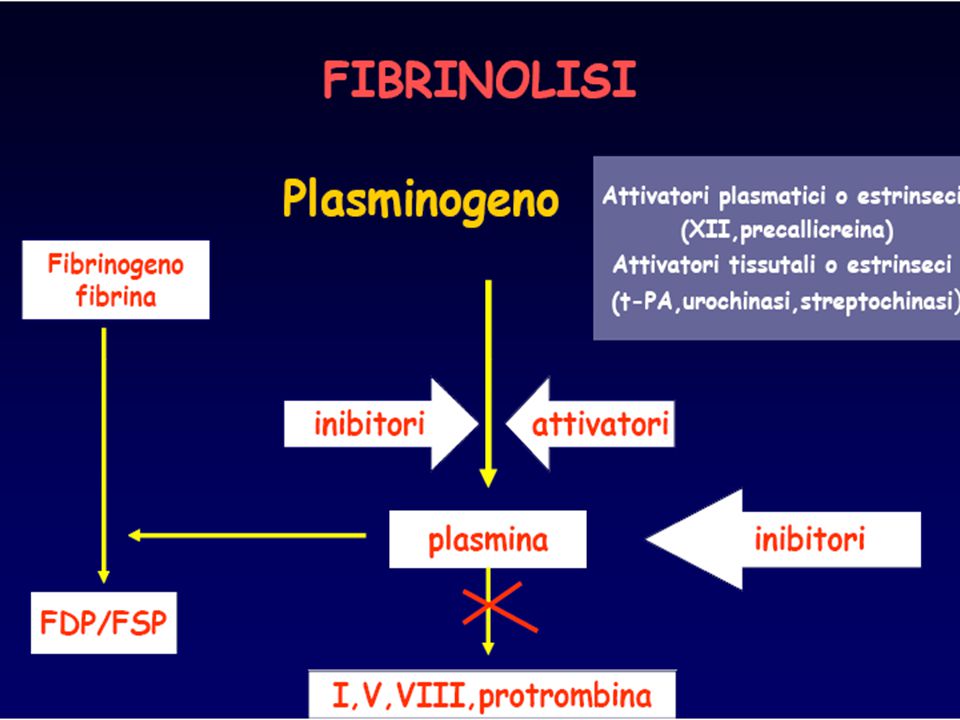
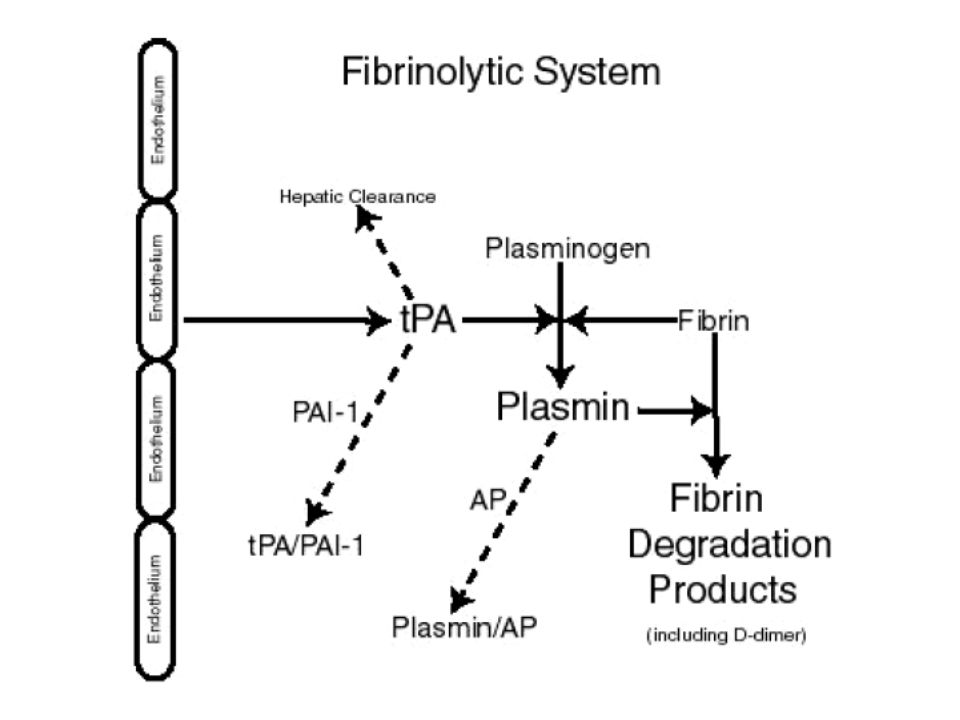
Tempo necessario alla formazione del coagulo di fibrina quando il plasma viene ricalcificato in presenza di sostanze come la cefalina e di attivatori della fase di contatto (acido ellagico, silice, caolino) Studia la via intrinseca e la via comune
 Studia la via intrinseca e la via comune.")
35
Tempo di tromboplastina parziale attivata (aPTT)
E’ sensibile alla presenza dei fattori della fase di contatto (precallicreina, chininogeno), ai fattori della via intrinseca (F XII, FXI, FIX, FVIII) e della via comune (FII, FV, FX, Fibrinogeno) E’ sensibile alla presenza di eparina E’ alterato in presenza di anticorpi antifosfolipidi (LAC e ACA)
, ai fattori della via intrinseca (F XII, FXI, FIX, FVIII) e della via comune (FII, FV, FX, Fibrinogeno) E’ sensibile alla presenza di eparina. E’ alterato in presenza di anticorpi antifosfolipidi (LAC e ACA)")
36
Tempo di protrombina Il plasma viene ricalcificato e in presenza di una tromboplastina e viene indotta la coagulazione Il tempo necessario alla formazione del coagulo in queste condizioni è il PT Studia la funzionalità della via estrinseca ed è sensibile ai fattori VII, V, X, protrombina e fibrinogeno E’ sensibile ai fattori vitamina K dipendenti (II, VII, IX ), quindi si usa per monitorare i farmaci antagonisti della vitamina K (anticoagulanti orali)
, quindi si usa per monitorare i farmaci antagonisti della vitamina K (anticoagulanti orali)")
37
Possibile espressione del PT
Velocità di coagulazione (secondi) Attività (percentuale) PT ratio (tra plasma del paziente e plasma di riferimento) INR…….vale solo per i pazienti in terapia anticoagulante orale!!!
 Attività (percentuale) PT ratio (tra plasma del paziente e plasma di riferimento) INR…….vale solo per i pazienti in terapia anticoagulante orale!!!")
38
Come leggere PT e aPTT L’espressione corretta dei risultati andrebbe fatta con un rapporto fra il tempo di coagulazione del campione e quello di un plasma pool di controllo Secondi plasma del paziente Secondi plasma pool = ratio PT o ratio aPTT L’ intervallo di riferimento può variare da lab a lab e andrebbe periodicamente rivalutato da parte del lab, testando il plasma pool di riferimento.

39
INR

40
INR Metodo di calcolo introdotto per ridurre la variabilità del tempo di coagulazione dipendente dai diversi reagenti impiegati (tromboplastine). Da usarsi solo per i pazienti anticoagulati con dicumarolici

43
I meccanismi di controllo della coagulazione

45
Antitrombina Scoperta nel 1905
Appartiene alla famiglia delle “serpine” (serin proteasi) Inibisce la trombina, FXa e in misura minore i FXIa e FXIIa, plasmina, callicreina e C1 (complemento) L’azione dipende dalla formazione di un complesso 1:1 tra il sito reattivo dell’AT e il dominio attivo delle proteine target L’effetto è potenziato in presenza dei glicosaminoglicani (GAG) esposti dall’endotelio o dall’eparina
 Inibisce la trombina, FXa e in misura minore i FXIa e FXIIa, plasmina, callicreina e C1 (complemento) L’azione dipende dalla formazione di un complesso 1:1 tra il sito reattivo dell’AT e il dominio attivo delle proteine target. L’effetto è potenziato in presenza dei glicosaminoglicani (GAG) esposti dall’endotelio o dall’eparina.")
46
…e se manca? Il difetto di AT è stata la prima causa di “trombofilia” congenita scoperta Nel 1965 Egeberg descrisse una famiglia norvegese con episodi recidivanti di trombosi venosa …alla prossima lezione maggiori dettagli

47
Proteina C Proteina C: scoperta agli inizi degli anni ’80.
Proteina di sintesi epatica, vitamina K dipendente E’ uno zimogeno: prodotto in forma inattiva, viene attivata dalla trombina Ha un’azione di inibizione sul FVa e sul FVIIIa

48
Proteina S Deve il suo nome a “Seattle” città dove fu scoperta nel 1984 Proteina vitamina K dipendente, di sintesi epatica Presente in circolo in due forme: una libera e attiva (circa il 40%) e una legata ad un residuo proteico, inattiva Ha azione di “cofattore” per la proteina C e ne potenzia l’attività
 e una legata ad un residuo proteico, inattiva. Ha azione di cofattore per la proteina C e ne potenzia l’attività.")
49
Protein C and Protein S

50
Patologie dell’emostasi
50

51
Un po’ di patologia dell’emostasi
51

52
Petecchie 52

53
53

54
Epistassi 54

55
Ematoma 55

56
? 56

57
Disordini congeniti ed acquisiti dell’emostasi
Malattia di Von Willebrand (vWD) Trombocitopenia autoimmune Piatsrinopenie secondarie (cirrosi, malattie autoimmuni, tumori, chemioterapia, farmaci vari) Emofilia e altri disordini emorragici congeniti (malattia di Glanzmann, malattia di Bernard-Soulier ) Anticorpi anti Antifosfolipidi (APA) 57
 Trombocitopenia autoimmune. Piatsrinopenie secondarie (cirrosi, malattie autoimmuni, tumori, chemioterapia, farmaci vari) Emofilia e altri disordini emorragici congeniti (malattia di Glanzmann, malattia di Bernard-Soulier ) Anticorpi anti Antifosfolipidi (APA) 57.")
58
Malattia di von Willebrand (vWD)
Nel 1926 Erik von Willebrand descrive il caso di una bimba finlandese di 5 anni affetta da diatesi emorragica importante Il pattern ereditario era differente dall’emofilia tradizionale, con carattere autosomico Nella stessa famiglia 23 soggetti su 66, di entrambi i sessi erano affetti, con sanguinamenti mucocutanei e tempo di emorragia prolungato 58

59
Malattia di von Willebrand (vWD)
Il più comune disordine emorragico ereditario. Prevalenza: circa 1-2 % La gravità della diatesi emorragica è proporzionale alla carenza di fattore von Willebrand (VWF) e alla carenza secondaria di fattore VIII (FVIII), poiché il VWF è la proteina carrier del FVIII nel plasma Trasmissione autosomica dominante e recessiva 59
 e alla carenza secondaria di fattore VIII (FVIII), poiché il VWF è la proteina carrier del FVIII nel plasma. Trasmissione autosomica dominante e recessiva. 59.")
60
60

61
Classification of von Willebrand disease
vW II N (Normandy) : livelli di FVIII basso con vW normale, per ridotta affinità della proteina per il FVIII dovuta a mutazione dle sito di legame con il FVIII. Nel WD 2B aumenta l’affinità del fattore willebrand x le plt. Tale anomalia determina legami spontanei dei multimeri ad alto peso molecolare del VWF alle piastrine, con conseguente rapida clearance dal plasma sia delle piastrine (aumento del rischio di trombocitopenia), che dei multimeri ad alto peso molecolare della VWF. La malattia si manifesta con sanguinamenti anomali a livello muco-cutaneo (menorragia, epistassi, emorragie gastrointestinali etc.). La malattia è causata da mutazioni nel gene VWF e la trasmissione è autosomica dominante. 61
 : livelli di FVIII basso con vW normale, per ridotta affinità della proteina per il FVIII dovuta a mutazione dle sito di legame con il FVIII. Nel WD 2B aumenta l’affinità del fattore willebrand x le plt. Tale anomalia determina legami spontanei dei multimeri ad alto peso molecolare del VWF alle piastrine, con conseguente rapida clearance dal plasma sia delle piastrine (aumento del rischio di trombocitopenia), che dei multimeri ad alto peso molecolare della VWF. La malattia si manifesta con sanguinamenti anomali a livello muco-cutaneo (menorragia, epistassi, emorragie gastrointestinali etc.). La malattia è causata da mutazioni nel gene VWF e la trasmissione è autosomica dominante. 61.")
62
Clinical characteristics of vWD
Both sexes are affected Mucocutaneous bleeding is the predominant symptom In women menhorragia may be the only manifestation Prolonged bleeding time with normal platelet count is the most important lab abnormality Soft tissue and joint bleeds are rare (except in type 3 vWD) 62
 62.")
63
How I treat VWD? General principle
The goal is to treat the dual defect of haemostasis: abnormal platelet adhesion due to low VWF and the abnormal intrinsic coagulation due to low FVIII Desmopressin: it releases endogenous VWF from endothelial cells Exogenous VWF contained in plasma derived concentrates 63

64
Haemophilia A & B Recessive inherited X-linked
Prevalence 1:5000 (A), 1:25000 (B) Severity: Mild -> FVIII 5-40% Moderate -> FVIII 1-5% Severe -> FVIII < 1% 64
, 1:25000 (B) Severity: Mild -> FVIII 5-40% Moderate -> FVIII 1-5% Severe -> FVIII < 1% 64.")
65
Clinica dell’emofilia
65

66
Clinical manifestation of haemophilia
Musculoskeletal bleeding: 85% in the joint, 15% muscles Bleeding can occur spontaneously or after minor trauma The first episode often occurs in the first 2 years of life when walking begins Women carriers can report menorrhagia, bleeding in pregnancy or labor 66

67
How I treat Haemophilia? General principle
The main treatment is to replace the clotting factor (FVIII or FIX) The prophylactic therapy is used to prevent bleeding Desmopressin: it releases endogenous FVIII from endothelial cells. Only for mild haemphilia Exogenous FVIII contained in plasma derived concentrates or recombinant concentrates 67
 The prophylactic therapy is used to prevent bleeding. Desmopressin: it releases endogenous FVIII from endothelial cells. Only for mild haemphilia. Exogenous FVIII contained in plasma derived concentrates or recombinant concentrates. 67.")
68
Tromboastenia di Glanzmann
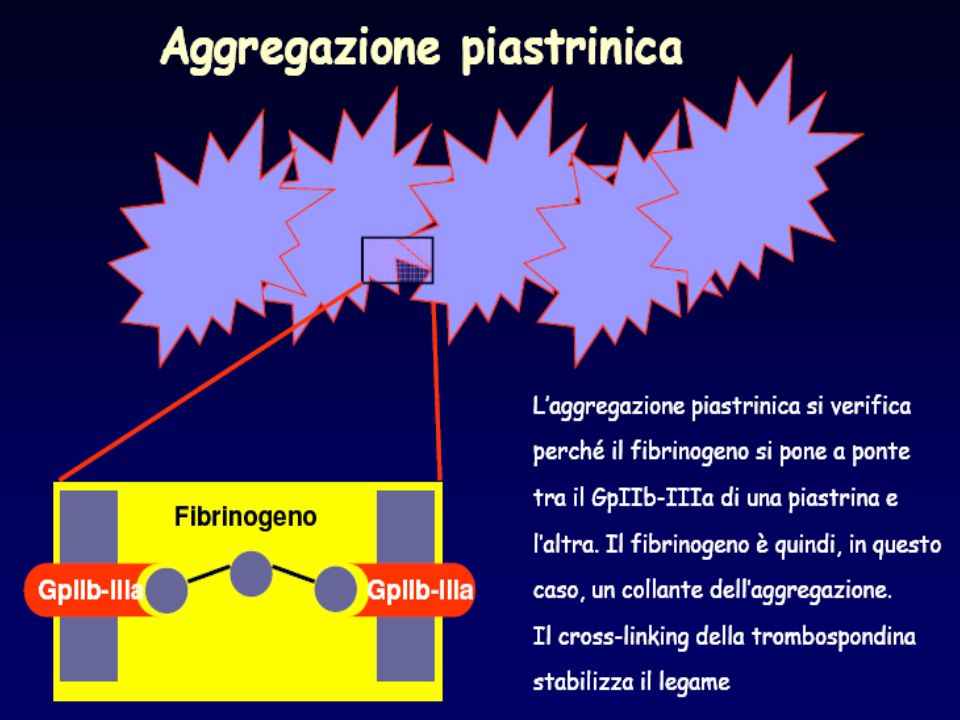
Disordine autosomico recessivo delle piastrine Prolungamento del tempo di emorragia Normale conta piastrinica Assente aggregazione a tutti gli agenti aggreganti legato alla carenza o disfunzione della GPIIb/IIIa (recettore per il fibrinogeno) 68
 68.")
69
Tromboastenia di Glanzmann
Eterozigoti asintomatici o con lieve diatesi emorragica mucocutanea Omozigoti: episodi emorragici anche gravi, epistassi, gengivorragia soprattutto dopo interventi odontoiatrici, emorragie post partum 69

70
Tromboastenia di Glanzmann
Tipo I: carenza quasi totale di GpIIb/IIIa (< 1%) Tipo II: carenza di GpIIb/IIIa con livelli 10-20% Tipo III: dosaggio quantitativo normale di GpIIb/IIIa ma proteina disfunzionale Diagnosi: con test funzionali e citofluorimetria 70
 Tipo II: carenza di GpIIb/IIIa con livelli 10-20% Tipo III: dosaggio quantitativo normale di GpIIb/IIIa ma proteina disfunzionale. Diagnosi: con test funzionali e citofluorimetria. 70.")
71
Trombofilia ereditaria
Condizione di aumentato rischio di sviluppo di eventi tromboembolici, solitamente venosi (ma possibili anche arteriosi) su base genetica Causata da difetti quantitativi o qualitativi delle proteine anticoagulanti naturali (AT, PC, PS) o da mutazioni genetiche di alcuni fattori della coagulazioni con conseguente ipercoagulabilità 71
 su base genetica. Causata da difetti quantitativi o qualitativi delle proteine anticoagulanti naturali (AT, PC, PS) o da mutazioni genetiche di alcuni fattori della coagulazioni con conseguente ipercoagulabilità. 71.")
72
Carenza di Antitrombina
La più rara: 1: 5000 Eventi di trombosi anche in giovane età Famigliarità per eventi tromboembolici Episodi recidivanti di trombosi Trombosi in gravidanza, puerperio o durante l’uso di anticoncezionali 72

73
Carenza di Antitrombina
Tipo I: carenza quantitativa. Forma “classica” con riduzione del dosaggio antigenico e funzionale del 50% circa Tipo II: alterazione qualitativa con livelli plasmatici quantitativi normali, ma deficit funzionale 73

74
Deficit di Proteina C & Proteina S
Carenza PC: in forma omozigote spesso incompatibile con la vita per comparsa di porpora fulminante alla nascita La carenza riduce la capacità di downregulation della generazione di trombina per attivazione da parte del FVa e FVIIIa Carenza PS: quantitativa o qualitativa. Causa una ridotta efficacia di azione della proteina C poiché funziona come suo cofattore 74

75
Activated protein C resistance
FVa resistance to activated Protein C Glu 506 FATTORE V Leiden 75

76
V Leiden Il Fattore Va Leiden mantiene inalterata la propria attività procoagulante, ma resiste alla inattivazione, determinando un aumentato rischio di trombosi Prevalenza variabile 3-13% nella popolazione generale, circa 10-20% nei soggetti con trombosi 76

77
G20210A protrombina La mutazione G20210 (sostituzione della Guanina con Adenosina in posizione del gene della protrombina) si esprime fenotipicamente con un aumento nel plasma della concentrazione di protrombina Prevalenza 2-12% nella popolazione generale 77
 si esprime fenotipicamente con un aumento nel plasma della concentrazione di protrombina. Prevalenza 2-12% nella popolazione generale. 77.")
78
Trombofilia ereditaria: principali cause identificate
Anno della scoperta Difetto di antitrombina Difetto di proteina C Difetto di proteina S Fattore VLeiden (G1691A) Mutazione protrombina (G20210A) 1996 (Disfibrinogenemia) < 10% degli eventi TEV 40% degli eventi 6 78
 Mutazione protrombina (G20210A) (Disfibrinogenemia) < 10% degli eventi TEV. 40% degli eventi")
79
Prevalenza trombofilia ereditaria
popolazione pazienti generale (unselected) deficit antitrombina % % deficit proteina C % % deficit protein S ? % fattore V Leiden % % protrombina G20210A % % 79
 deficit antitrombina % 1 % deficit proteina C % 3 % deficit protein S % fattore V Leiden % % protrombina G20210A % % 79.")
80
Rischio di TEV e trombofilia ereditaria
aumento rischio relativo antitrombina proteina C proteina S fattore V Leiden protrombina G20210A Aggiungere la diapo con le prevalenze? Prevalenza di PS 1:30000 pop gen, 2-3% pz unselected 80

81
Le alterazioni iatrogene dell’emostasi
81

82
82

83
Available Drugs as Antithrombotic therapy
Antiplatelet drugs (aspirin, clopidogrel, ticlopidin) Heparin: unfractionated heparin, low molecular weight heparin Oral Anticoagulant: vitamin K antagonists New anticoagulant (oral or parenteral) 83
 Heparin: unfractionated heparin, low molecular weight heparin. Oral Anticoagulant: vitamin K antagonists. New anticoagulant (oral or parenteral) 83.")
84
Mode of Action of Antiplatelet Agents
CLOPIDOGREL ASA COX ADP C GPllb/llla (Fibrinogen receptor) Collagen thrombin TXA 2 Activation This slide illustrates the pathways involved in platelet activation and the points at which currently available anti-platelet therapies act:1 Clopidogrel is a specifc, non-competive inhibitor of adenosine diphosphate- (ADP) induced platelet aggregation, irreversibly inhibiting the binding of ADP to its platelet membrane receptors. Ultimately it inhibits the activation of the GPIIb/IIIa receptor, its binding to fibrinogen and further platelet aggregation. ASA affects the thromboxane A2 (TXA2) pathway and is the most widely used antiplatelet treatment. ASA inhibits the cyclo-oxygenase enzyme, reducing the production of prostaglandin and TXA2 from arachidonic acid. TXA2 activates the GPIIb/IIIa binding site on the platelet, allowing fibrinogen to bind. Dipyridamole inhibits platelet aggregation by blocking the reuptake of adenosine formed from precursors released by red blood cells following microtrauma. GPIIb/IIIa blockers are competitive inhibitors of the glycoprotein (GP)IIb/IIIa receptor sites found on the surface of activated platelets. They inhibit platelet aggregation by preventing the binding of fibrinogen, von Willebrand factor and other adhesive ligands to GPIIb/IIIa receptors. COX (cyclo-oxygenase) ADP (adenosine diphosphate) TXA2 (thromboxane A2) Schafer AI. Am J Med 1996; 101: 199–209. Reference: 1. Schafer AI. Am J Med 1996; 101: 199–209. 84
 Collagen thrombin. TXA. 2. Activation. This slide illustrates the pathways involved in platelet activation and the points at which currently available anti-platelet therapies act:1. Clopidogrel is a specifc, non-competive inhibitor of adenosine diphosphate- (ADP) induced platelet aggregation, irreversibly inhibiting the binding of ADP to its platelet membrane receptors. Ultimately it inhibits the activation of the GPIIb/IIIa receptor, its binding to fibrinogen and further platelet aggregation. ASA affects the thromboxane A2 (TXA2) pathway and is the most widely used antiplatelet treatment. ASA inhibits the cyclo-oxygenase enzyme, reducing the production of prostaglandin and TXA2 from arachidonic acid. TXA2 activates the GPIIb/IIIa binding site on the platelet, allowing fibrinogen to bind. Dipyridamole inhibits platelet aggregation by blocking the reuptake of adenosine formed from precursors released by red blood cells following microtrauma. GPIIb/IIIa blockers are competitive inhibitors of the glycoprotein (GP)IIb/IIIa receptor sites found on the surface of activated platelets. They inhibit platelet aggregation by preventing the binding of fibrinogen, von Willebrand factor and other adhesive ligands to GPIIb/IIIa receptors. COX (cyclo-oxygenase) ADP (adenosine diphosphate) TXA2 (thromboxane A2) Schafer AI. Am J Med 1996; 101: 199–209. Reference: 1. Schafer AI. Am J Med 1996; 101: 199–")
85
Aspirina il farmaco acetila la COX1 delle piastrine in modo irreversibile a livello della serina 529 presente sul sito attivo dell’enzima. 85

86
Acido acetisalicilico (ASA)
Inibisce irreversibilmente la COX-1 piastrinica L’effetto è presente già dopo 60’ dall’assunzione del farmaco e persiste per giorni 86

87
Effetti collaterali dell’ASA
Emorragia G.I. con rischio dose-dipendente, presente anche a dosi di mg/die Rischio di 2-3 volte rispetto a non ASA (parzialmente ridotto con l’uso di gastroprotettori) Incidenza annua è di 1-2 /1000 pts trattati (con l’età, helicobacter pylori, altri FANS) Emorragie cerebrali (< 1/1000 pts trattati, ipertensione arteriosa non controllata) Nel paziente anziano è importante valutare il rischio/beneficio 87
 Incidenza annua è di 1-2 /1000 pts trattati (con l’età, helicobacter pylori, altri FANS) Emorragie cerebrali (< 1/1000 pts trattati, ipertensione arteriosa non controllata) Nel paziente anziano è importante valutare il rischio/beneficio. 87.")
88
Tienopiridine (ticlopidina, clopidogrel)
Azione irreversibile, inibiscono il recettore P2Y12, delle piastrine. Ambedue le sostanze sono pro-farmaci richiedono il metabolismo epatico mediato dal citocromo P450 isoforma 3A4. Si forma un metabolita attivo che agisce sul recettore piastrinico. Slow onset of action (metabolismo epatico) Somministrazione di una “loading dose” (300mg Clopidogrel) prima delle procedure acute (PCI) 88
 Somministrazione di una loading dose (300mg Clopidogrel) prima delle procedure acute (PCI) 88.")
89
Farmaci antiaggreganti:Tienopiridine
Ticlopidina e Clopidogrel Inibizione selettiva ed irreversibile per recettore piastrinico dell’ADP Sono efficaci in vivo dopo trasformazione in metaboliti attivi (attraverso citocromo P450) L’azione è lenta (2-3 giorni) Miglior profilo tollerabilità gastrica Effetti collaterali Neutropenia (3%), porpora trombotica trombocitopenica (0,02% >50 volte rispetto alla popolazione generale), diarrea, rush cutanei, aumento colesterolo Clopidogrel: farmacocinetica migliore, minor effetti collaterali 89
 L’azione è lenta (2-3 giorni) Miglior profilo tollerabilità gastrica. Effetti collaterali. Neutropenia (3%), porpora trombotica trombocitopenica (0,02% >50 volte rispetto alla popolazione generale), diarrea, rush cutanei, aumento colesterolo. Clopidogrel: farmacocinetica migliore, minor effetti collaterali. 89.")
90
A brief history of anticoagulant therapy
Unfractionated heparins: antithrombin (AT)-dependent inhibition of Factor Xa and IIa in a 1:1 ratio 1930s Parenteral Vitamin K antagonists:indirectly affect synthesis of multiple coagulation factors 1940s Oral Low molecular weight heparins: AT-dependent inhibition of factor Xa and IIa Parenteral 1980s Oral Direct Factor IIa inhibitors 1990s Parenteral Indirect Factor Xa inhibitors 2000s Direct Factor IIa inhibitors Direct Factor Xa inhibitors Oral today Alban, Eur J Clin Invest 2005; Link, Circulation 1959 90
-dependent inhibition of Factor Xa and IIa in a 1:1 ratio. 1930s. Parenteral. Vitamin K antagonists:indirectly affect synthesis of multiple coagulation factors. 1940s. Oral. Low molecular weight heparins: AT-dependent inhibition of factor Xa and IIa. Parenteral. 1980s. Oral. Direct Factor IIa inhibitors. 1990s. Parenteral. Indirect Factor Xa inhibitors. 2000s. Direct Factor IIa inhibitors. Direct Factor Xa inhibitors. Oral. today. Alban, Eur J Clin Invest 2005; Link, Circulation")
91
Eparina Eparina: glicosaminoglicano
Elevata affinità per l’Antitrombina III (potente inibitore naturale della coagulazione) Eparina non frazionata Si lega a molte proteine plasmatiche, metabolismo epatico-renale, (clearance rapida) Effetti collaterali: emorragia (2-5%), rischio con aumentato dosaggio, età paziente (>70anni), trombocitopenia, osteoporosi Monitoraggio: APTT (1,5-2,5) volte il valore basale 91
 Eparina non frazionata. Si lega a molte proteine plasmatiche, metabolismo epatico-renale, (clearance rapida) Effetti collaterali: emorragia (2-5%), rischio con aumentato dosaggio, età paziente (>70anni), trombocitopenia, osteoporosi. Monitoraggio: APTT (1,5-2,5) volte il valore basale. 91.")
92
Eparina a basso peso molecolare:
Maggiore biodisponibilità Caratteristiche farmacocinetiche migliori Risposta antitrombotica prevedibile Emivita più lunga Minor rischio emorragico (1-2%) Non monitoraggio di laboratorio 92
 Non monitoraggio di laboratorio. 92.")
93
Anticoagulanti orali Dicumarolici:
derivati chimici delle 4-idrossicumarina In Italia: Warfarin (cp da 5mg) acenocumarolo (Sintrom)dose da 4mg e 1mg Diversa emivita biologica Warfarin h Acenocumarolo h 93
 acenocumarolo (Sintrom)dose da 4mg e 1mg. Diversa emivita biologica. Warfarin 30-60h. Acenocumarolo 12h. 93.")
94
Vantaggi e svantaggi dei due farmaci
WARFARIN Trattamento a lunga durata, maggiore stabilità del range terapeutico Acenocumarolo Maggior reversibilità dell’effetto anticoagulante, 1mg utile in pazienti anziani AZIONE: a livello epatico diminuisce la sintesi fattori K dipendente (II-VII-IX-X) + Prot C-S 4-5 giorni per efficacia ottimale Risposta dipende (età, peso corporeo, compliance pts, assunzione altri farmaci, dieta) MONITORAGGIO DI LABORATORIO 94
 + Prot C-S. 4-5 giorni per efficacia ottimale. Risposta dipende (età, peso corporeo, compliance pts, assunzione altri farmaci, dieta) MONITORAGGIO DI LABORATORIO. 94.")
95
Dicumarolici Complicanza: emorragia Studio ISCOAT
Incidenza annuale 1-2% Fattori coinvolti: età, rischio >2 volte per pazienti >70y Livelli scoagulazione (INR>4,5, rischio > 6 volte i primi 90 giorni di terapia Indicazione alla terapia (vasculopatia arteriosa, storia ictus cerebrali, comorbidità epatica-renale) Necrosi cutanea (pts con deficit conferito di Prot-Prot S e/o sindrome da Ab antifosfolipidi Eritemi cutanei Alopecia 95
 Necrosi cutanea (pts con deficit conferito di Prot-Prot S e/o sindrome da Ab antifosfolipidi. Eritemi cutanei. Alopecia. 95.")
96
Gli anticoagulanti orali (1)
I farmaci anticoagulanti orali (AO) attualmente in uso (warfarina e acenocumarolo) sono molecole derivate dalla 4-idrossicumarina (dicumarolo). 96
 attualmente in uso (warfarina e acenocumarolo) sono molecole derivate dalla 4-idrossicumarina (dicumarolo). 96.")
97
Gli anticoagulanti orali (2)
L’attività anticoagulante dei derivati cumarinici è legata alla presenza del residuo 4-idrossicumarinico e di un atomo di carbonio sostituente in posizione 3. 97

98
Meccanismo d’azione degli AO (1)
I dicumarolici sono antagonisti della vitamina K e agiscono interferendo con la modificazione post-traslazionale epatica dei fattori della coagulazione vitamina K dipendenti (II, VII, IX, X, ma anche PC, PS). La vit.K è un cofattore essenziale per la carbossilazione dei residui di acido glutammico della regione N-terminale di questi fattori della coagulazione, dando origine ad un residuo aminoacidico γ-carbossiglutammico. 98
. La vit.K è un cofattore essenziale per la carbossilazione dei residui di acido glutammico della regione N-terminale di questi fattori della coagulazione, dando origine ad un residuo aminoacidico γ-carbossiglutammico. 98.")
99
Meccanismo d’azione degli AO (2)
I residui γ-carbossiglutammati delle proteine sintetizzate, sono responsabili della chelazione del calcio. La chelazione del calcio è necessaria affinchè i fattori della coagulazione vit.K dipendenti si leghino alle membrane cellulari contenenti fosfolipidi, sulle quali si attiva la cascata della coagulazione. 99

100
Variabilità della risposta agli AO
Fattori farmacocinetici e farmacodinamici: interazione con farmaci che ne modificano l’assorbimento o il metabolismo, abuso di alcol, contenuto di vitamina K nella dieta, epatopatia, resistenza geneticamente determinata Fattori esterni: inaccuratezza dei test, scarsa compliance del paziente, mancanza di comunicazione medico-paziente 100

101
Cause di variazione INR
101

102
Farmaci che potenziano l’effetto degli AO
amiodarone eritromicina cimetidina chinidina isoniazide omeprazolo propranololo metronidazolo propafenone fluconazolo danazolo cotrimossazolo tamoxifen ciprofloxacina simvastatina sulfinpirazone fibrati 102

103
Farmaci che riducono l’effetto degli AO
rifampicina ciclosporina nafcillina vitamina K carbamazepina spironolattone fenobarbitale estrogeni e progestinici fenintoina aloperidolo sucralfato colestiramina antiacidi 103

104
Characteristics of the ideal anticoagulant
Highly effective in reducing thromboembolic risk Low rate of bleeding events Administered orally, once daily Predictable dose response and kinetic No routine monitoring of coagulation Wide therapeutic window No dose adjustment required Little interaction with food or drugs Low non specific plasma protein binding Inhibition of both free and clot-bound activated coagulation factors Simple cheap antidote 104

105
Alternative Anticoagulants
ORAL PARENTERAL TF/VIIa TFPI (tifacogin) TTP889 X IX Rivaroxaban Apixaban LY517717 YM150 DU-176b Betrixaban TAK 442 APC (drotrecogin alfa) sTM (ART-123) IXa VIIIa Va AT Xa Fondaparinux Idraparinux Idrabiotaparinux There are many targets for novel anticoagulants in the coagulation pathway: Tissue factor pathway inhibitor (TFPI) bound to Factor Xa inactivates the tissue factor (TF)–Factor VIIa complex, preventing initiation of coagulation Activated protein C (APC) degrades Factors Va and VIIIa, and thrombomodulin (soluble; sTM) converts thrombin (Factor IIa) from a procoagulant to a potent activator of protein C Fondaparinux and idraparinux indirectly inhibit Factor Xa, requiring antithrombin (AT) as a cofactor Direct (AT-independent) inhibitors of Factor Xa include rivaroxaban (BAY 597939), LY517717, YM150 and DU-176b (all orally available), and DX-9065a (intravenous) Oral, direct thrombin inhibitors include ximelagatran (now withdrawn) and dabigatran Weitz JI & Bates SM. New anticoagulants. J Thromb Haemost 2005;3:1843–1853 Ximelagatran Dabigatran Argatroban Hirudin Lepidurin Bivalirudin II DX-9065a IIa Fibrinogen Fibrin Adapted from Weitz & Bates, J Thromb Haemost 2007 105
 TTP889. X. IX. Rivaroxaban Apixaban. LY YM150. DU-176b. Betrixaban. TAK 442. APC (drotrecogin alfa) sTM (ART-123) IXa. VIIIa. Va. AT. Xa. Fondaparinux Idraparinux. Idrabiotaparinux. There are many targets for novel anticoagulants in the coagulation pathway: Tissue factor pathway inhibitor (TFPI) bound to Factor Xa inactivates the tissue factor (TF)–Factor VIIa complex, preventing initiation of coagulation. Activated protein C (APC) degrades Factors Va and VIIIa, and thrombomodulin (soluble; sTM) converts thrombin (Factor IIa) from a procoagulant to a potent activator of protein C. Fondaparinux and idraparinux indirectly inhibit Factor Xa, requiring antithrombin (AT) as a cofactor. Direct (AT-independent) inhibitors of Factor Xa include rivaroxaban (BAY 597939), LY517717, YM150 and DU-176b (all orally available), and DX-9065a (intravenous) Oral, direct thrombin inhibitors include ximelagatran (now withdrawn) and dabigatran. Weitz JI & Bates SM. New anticoagulants. J Thromb Haemost 2005;3:1843–1853. Ximelagatran Dabigatran. Argatroban. Hirudin. Lepidurin. Bivalirudin. II. DX-9065a. IIa. Fibrinogen. Fibrin. Adapted from Weitz & Bates, J Thromb Haemost")
106
ITP: definizioni e nuovi scenari terapeutici
106

107
The panel decided to avoid the term “idiopathic,” preferring
“immune,” to emphasize the immune-mediated mechanism of the disease and to choose “primary” (as opposed to idiopathic) to indicate the absence of any obvious initiating and/or underlying cause. 107
 to. indicate the absence of any obvious initiating and/or underlying. cause")
108
Primary vs secondary immune thrombocytopenia
SLE, systemic lupus erythematosus; APS, antiphospholipid syndrome; CVID, common variable immune deficiency; CLL, chronic lymphocytic leukemia; APLS, autoimmune lymphoproliferative syndrome; post-tx, post-bone marrow or solid organ transplantation 108

109
ITP: phases of the disease

110
Diagnostic evaluation in suspected ITP

111
Examples of differential diagnosis of ITP identifies by patient history

112
Who should be treated?

113
Consensus recommendation for target platelet counts during surgery
Br J Haematol 2003

114
First-line therapies for ITP treatment

115
Treatment of unresponsive or relapsed ITP

116
Treatment of refractory ITP in adults (patients that have failed splenectomy or have relapsed thereafter)
")
117
Recommendations for treatment of ITP in pregnancy

Presentazioni simili