Scaricare la presentazione
1
Ematologia-Oncologia pediatrica
ANEMIE EMOLITICHE

2
Emolisi Definizione: riduzione della durata di vita degli eritrociti. Se l’entità della distruzione dei globuli rossi supera la capacità del midollo osseo a produrre più eritrociti, il disordine si manifesta come anemia emolitica.

3
Dimostrazione laboratoristica di processo emolitico
Aumentati livelli di bilirubinemia indiretta reticolocitosi esame della morfologia eritrocitaria (presenza di sferociti, ellissociti, etc.) Ulteriori conferme: Aptoglobina LDH Hb plasmatica Emivita GR (51Cr < 26 gg)
 Ulteriori conferme: Aptoglobina. LDH. Hb plasmatica. Emivita GR (51Cr < 26 gg)")
4
Le anemie emolitiche posso essere dovute a:
1. difetto intracorpuscolare 2. difetto extracorpuscolare Per il primo gruppo si valuteranno: le resistenze osmotiche il test dell’autoemolisi il test di Ham il test al calore o all’isopropanolo per la ricerca di emoglobine instabili le attività enzimatiche intraeritrocitarie Per il secondo gruppo si valuteranno: il test di Coombs la ricerca delle crioagglutinine agenti fisici e chimici

5
Anemie emolitiche da difetti della membrana eritrocitaria
Comprendono un gruppo importante di anemie ereditarie. I disordini più comuni sono: Sferocitosi ereditaria Ellissocitosi ereditaria

7
Sferocitosi ereditaria
È la più comune anemia ereditaria nelle popolazioni dell’Europa settentrionale, con un’incidenza di un caso su 2500 persone (ma, secondo alcuni, l’incidenza sarebbe ancora più elevata) nel 75% delle famiglie la malattia viene trasmessa con meccanismo autosomico dominante nel restante 25% dei casi si parla di forme “non dominanti”: in questo gruppo sono comprese forme recessive vere, e forme conseguenti a mutazioni “de novo”.
 nel 75% delle famiglie la malattia viene trasmessa con meccanismo autosomico dominante. nel restante 25% dei casi si parla di forme non dominanti : in questo gruppo sono comprese forme recessive vere, e forme conseguenti a mutazioni de novo .")
8
Le alterazioni della membrana eritrocitaria, che sono alla base della malattia, sono state suddivise in quattro gruppi principali: carenza combinata di anchirina e spectrina carenza isolata di spectrina carenza isolata di proteina di banda 3 carenza isolata di proteina 4.2 Le ricerche condotte negli ultimi anni hanno consentito di clonare i geni delle varie proteine coinvolte e di individuare in molti casi le mutazioni responsabili.

9
Carenza combinata di anchirina e spectrina
È il fenotipo biochimico più comune (60-65% dei casi). Il difetto primitivo interessa l’anchirina; la carenza di spectrina è secondaria e proporzionale a quella di anchirina.
. Il difetto primitivo interessa l’anchirina; la carenza di spectrina è secondaria e proporzionale a quella di anchirina.")
11
Carenza isolata di spectrina
È il gruppo meno comune. Sono state riscontrate alterazioni che coinvolgono sia la catena b che la catena a: nel primo caso si tratta di forme dominanti, nel secondo di forme recessive, poiché la sintesi di catene a nelle cellule normali è di 3-4 volte superiore a quella delle catene b.

12
Carenza isolata di proteina di banda 3
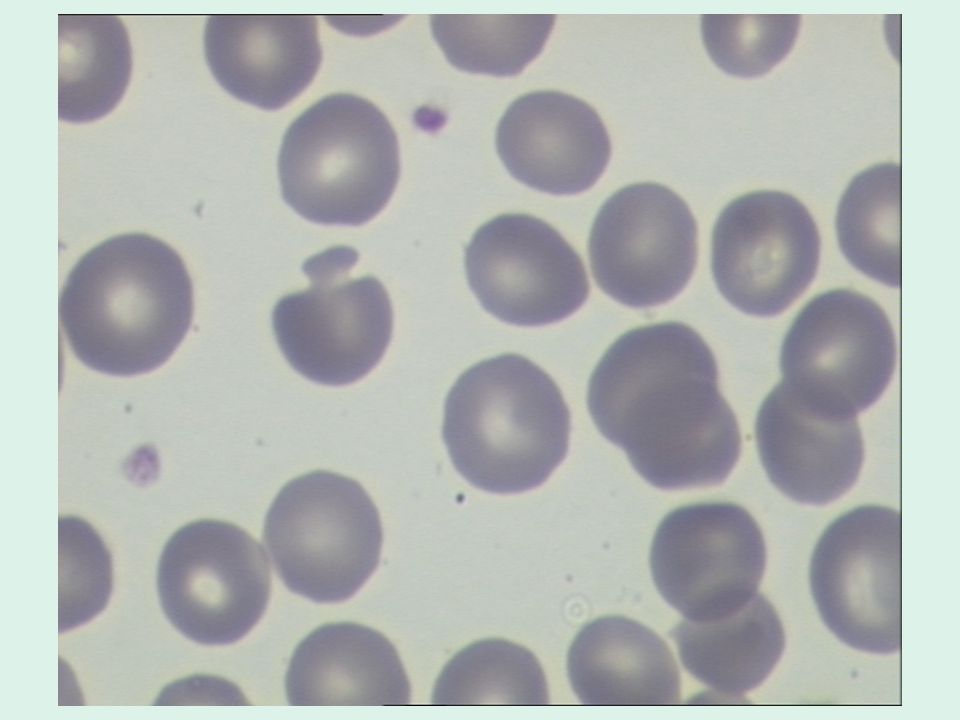
Questo gruppo comprende circa il 20-25% di tutti i casi di sferocitosi ereditaria. Si tratta di forme con fenotipo clinico lieve o moderato e presenza, all’osservazione dello striscio, di sferociti “pinzettati” o “a fungo”.

14
Carenza isolata di proteina 4.2
È un gruppo relativamente raro; la maggior parte dei casi sono stati descritti in Giappone. Si tratta di forme recessive, caratterizzate spesso, all’osservazione dello striscio, da ovalo-sferocitosi.

15
Fisiopatologia Le alterazioni proteiche descritte determinano
Disorganizzazione della struttura della membrana cellulare e perdita conseguente del doppio strato lipidico Riduzione del rapporto superficie/volume dei globuli rossi

16
Presenza in circolo di microsferociti, dotati di minore deformabilità
Aumentata fragilità osmotica dei globuli rossi Presenza in circolo di microsferociti, dotati di minore deformabilità Difficoltà ad attraversare le strette fenestrature della parete dei sinusoidi splenici “condizionamento splenico”

17
Ulteriore danno a carico della membrana cellulare
Fagocitosi dei globuli rossi danneggiati da parte dei macrofagi splenici Emolisi cronica extravascolare

18
Clinica della sferocitosi ereditaria
I segni clinici distintivi sono rappresentati da: Anemia, usualmente normocronica e normocitica ittero, a bilirubinemia prevalentemente indiretta splenomegalia Il quadro clinico è estremamente eterogeneo, in dipendenza di vari fattori quali: tipo di difetto biochimico, età, etc.

19
Classificazione clinica della sferocitosi ereditaria
Portatori di sferocitosi ereditaria Sono soggetti con fenotipo clinico ed ematologico pressoché normale, individuati per lo più nel corso di studi familiari. Presentano solo lievi alterazioni dei dati di laboratorio, quali modico aumento dei reticolociti e/o riduzione dell’aptoglobina e/o fragilità osmotica lievemente aumentata. Sferocitosi ereditaria lieve Rappresenta il 20-30% dei casi. Si tratta di pazienti con evidenza laboratoristica di emolisi, ben compensata (Hb normale), individuati spesso nel corso di studi familiari e/o per l’insorgenza di complicanze della malattia (es. litiasi biliare, crisi ipermolitiche, etc.)
, individuati spesso nel corso di studi familiari e/o per l’insorgenza di complicanze della malattia (es. litiasi biliare, crisi ipermolitiche, etc.)")
20
Sferocitosi ereditaria moderata
Rappresenta il gruppo più numeroso. Questi pazienti presentano il quadro clinico, ematologico e laboratoristico tipico della malattia, con emolisi non completamente compensata, e modica anemia (Hb: 8-12 gr/dl). Sferocitosi ereditaria moderatamente severa In questo gruppo rientrano i soggetti con anemia più marcata (Hb: 6-8 gr/dl). Sferocitosi ereditaria severa Rappresenta il gruppo meno numeroso (< 5% dei casi). Sono pazienti con anemia grave (Hb < 6 gr/dl) trasfusione-dipendenti; rappresentano le vere e proprie forme recessive della malattia.
. Sferocitosi ereditaria moderatamente severa. In questo gruppo rientrano i soggetti con anemia più marcata (Hb: 6-8 gr/dl). Sferocitosi ereditaria severa. Rappresenta il gruppo meno numeroso (< 5% dei casi). Sono pazienti con anemia grave (Hb < 6 gr/dl) trasfusione-dipendenti; rappresentano le vere e proprie forme recessive della malattia.")
21
Diagnosi di sferocitosi ereditaria
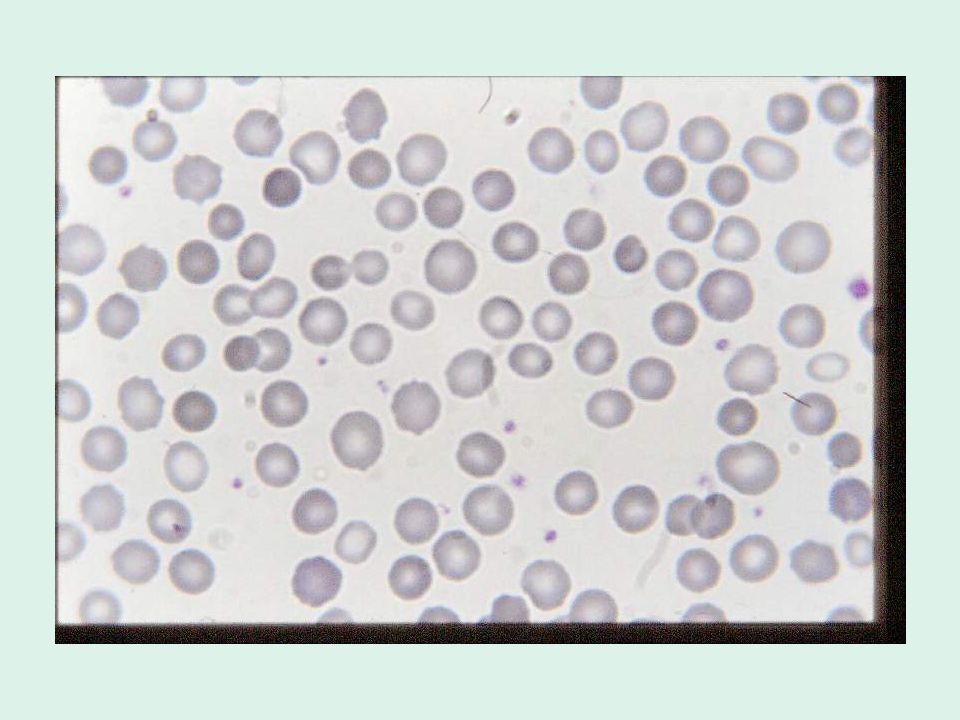
1. storia familiare positiva 2. esame clinico 3. evidenza laboratoristica di emolisi 4. presenza in circolo di microsferociti rilevabili: a. mediante osservazione dello striscio periferico b. mediante citometria da flusso: percentuale di cellule ipercromiche/ microcitiche-normocitiche valutazione MCV/MSCV

22
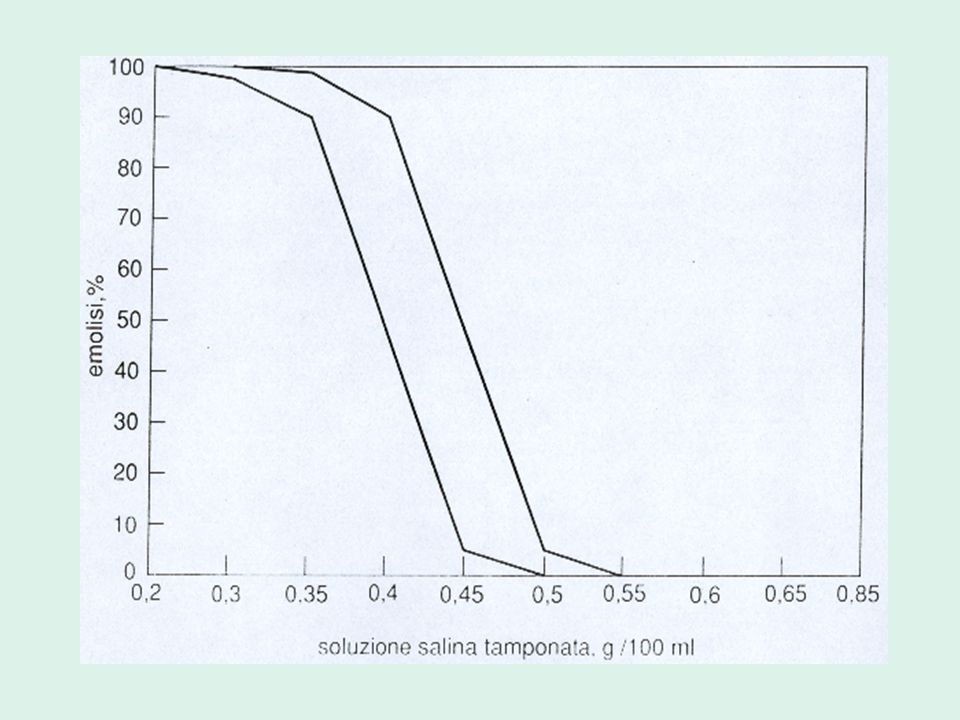
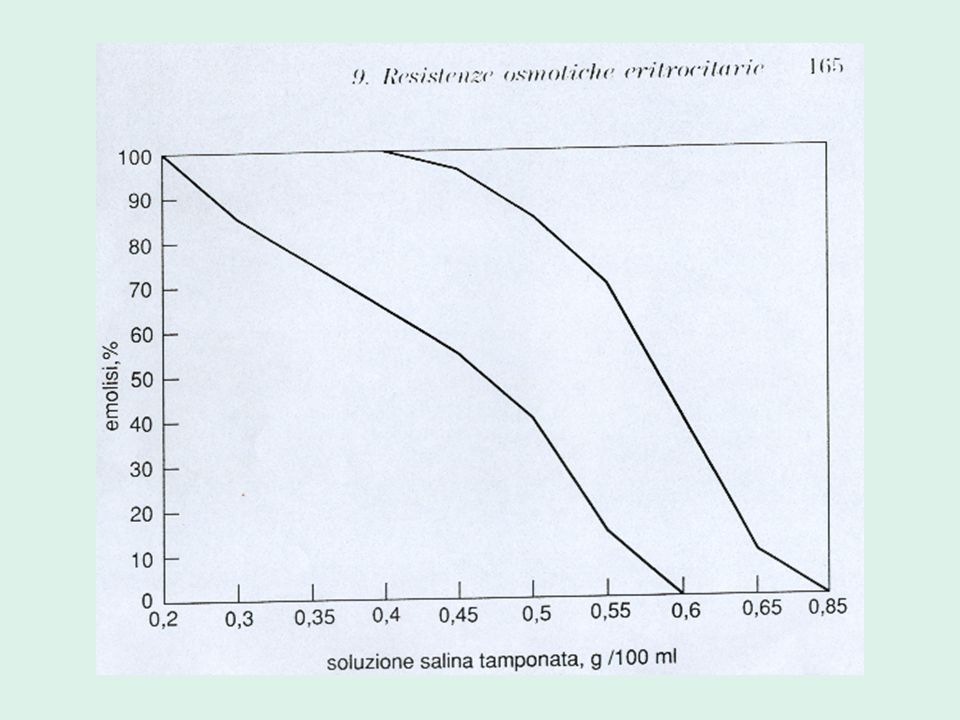
Valutazione delle resistenze osmotiche eritrocitarie
5. aumentata fragilità osmotica dei globuli rossi Valutazione delle resistenze osmotiche eritrocitarie Metodo Dacie-Parpart Test al glicerolo

23
RESISTENZE OSMOTICHE ERITROCITARIE
Valutano la fragilità delle emazie nei confronti di soluzioni saline ipotoniche

24
I GLOBULI ROSSI: IMMERSI IN UNA SOLUZIONE SALINA FISIOLOGICA:
mantengono la loro forma SOSPESI IN ACQUA DISTILLATA: Emolizzano rapidamente

25
Le resistenze osmotiche eritrocitarie dipendono da:
-FORMA -VOLUME -SUPERFICIE

26
Nella sferocitosi ereditaria:
Le emazie presentano un rapporto superficie-volume ridotto: è sufficiente l’ingresso di poca acqua perché tali cellule si rompano. Le resistenze osmotiche sono diminuite, ovvero la loro fragilità osmotica è aumentata.

27
Metodo Parpart Valuta l’entità dell’emolisi di sospensione di globuli rossi in soluzioni di NaCl progressivamente più ipotoniche. Si esegue: Sangue fresco Sangue incubato a 37° per 24 h sterilmente

30
Tempo di lisi al glicerolo (GLT)
Si utilizza un’unica soluzione di NaCl a cui è aggiunto glicerolo (come mezzo per ritardare l’emolisi) I risultati vengono espressi come tempo di dimezzamento della densità ottica del campione dei globuli rossi in esame (GLT50)
 I risultati vengono espressi come tempo di dimezzamento della densità ottica del campione dei globuli rossi in esame (GLT50)")
31
Modalità di esecuzione:
Un solo ml di sangue anticoagulato con EDTA Non è richiesta la sterilità

32
Test di lisi al glicerolo acidificato (AGLT)
Il Ph della soluzione lisante è più basso I risultati sono espressi come tempo di dimezzamento della densità ottica del campione in esame (AGLT50)
")
33
Modalità di esecuzione:
Con 20 ul di sangue Non è necessaria la sterilità Procedimento rapido e semplice

34
Pink- test Modifica della soluzione lisante per stabilizzare il Ph
I risultati sono espressi come percentuale di emolisi del campione in esame,rispetto alla completa emolisi del campione stesso. Maggiore rapidità e semplicità

35
Osmored 2 reattivi A e B vengono forniti già preparati per la misura del grado di iper-resistenza e del grado di iporesistenza Si aggiunge poi una soluzione lisante I campioni normali presentano una lisi in provetta A>70% I campioni normali presentano una lisi in provetta B<30%

36
Diagnosi differenziale
Va fatta con tutte le condizioni morbose per lo più acquisite, caratterizzate da anemia emolitica sferocitica, come ad es. l’incompatibilità neonatale ABO, l’anemia emolitica autoimmune, etc. La valutazione di tutti gli elementi diagnostici precedentemente descritti, unitamente all’esecuzione di pochi tests (ad es. determinazione dei gruppi sanguigni, test di Coombs diretto, etc.), consentono di porre, abbastanza agevolmente, la diagnosi.
, consentono di porre, abbastanza agevolmente, la diagnosi.")
37
Complicanze Litiasi biliare: è la più frequente complicanza della sferocitosi ereditaria. Come in altre anemie emolitiche, la coeredità di ittero di Gilbert e sferocitosi ereditaria determina un’insorgenza più precoce e più frequente di tale complicanza. Crisi emolitiche: in genere scatenate da infezioni, per lo più virali, sono caratterizzate da un’accentuazione, in genere lieve, dell’emolisi e conseguente peggioramento dell’anemia. Crisi aplastiche: comuni ad altre anemie emolitiche croniche, sono dovute all’infezioni da Parvovirus B19, agente etiologico dell’eritema infettivo o V malattia. Crisi megaloblastiche: l’accentuazione dell’anemia è conseguenza della carenza di acido folico.

38
Terapia Somministrazione di acido folico Emotrasfusioni Splenectomia

39
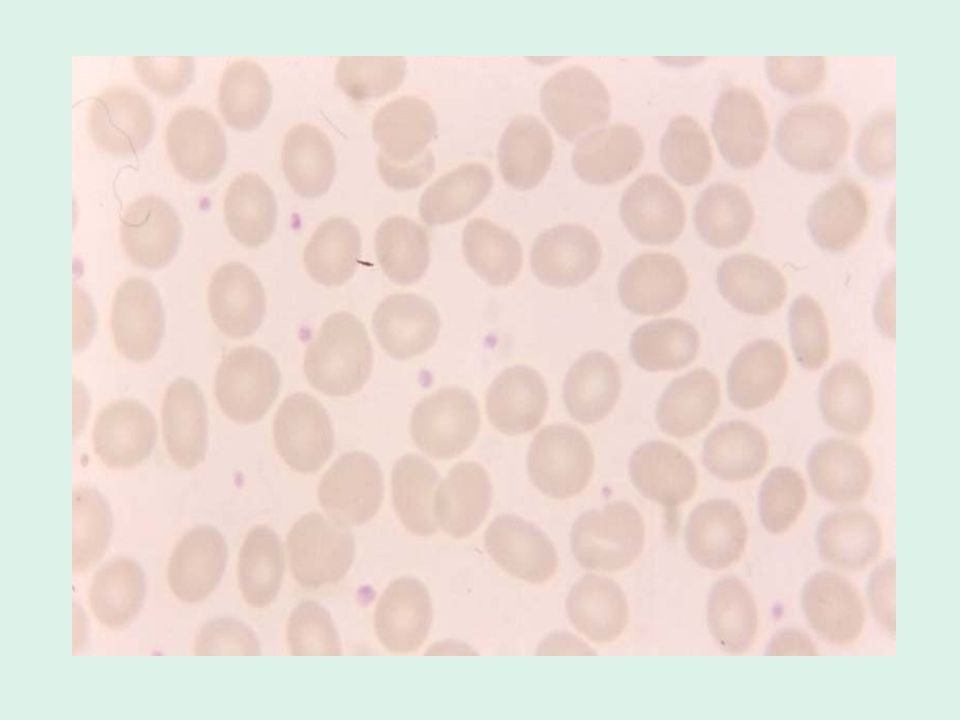
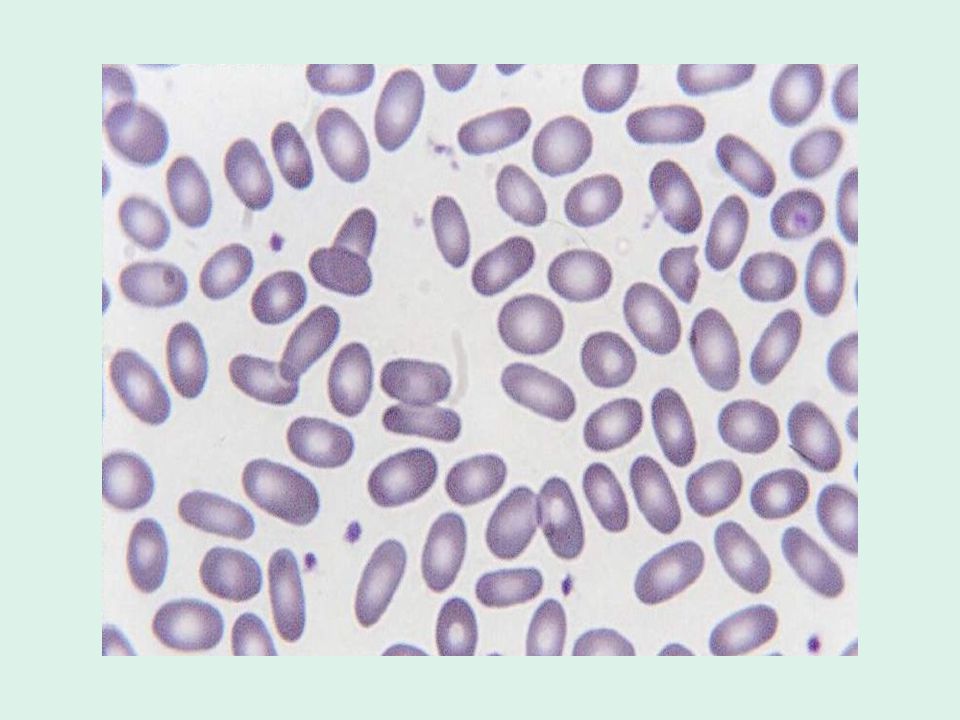
Ellissocitosi ereditaria
È caratterizzata dalla presenza di eritrociti ellittici o ovali all’osservazione dello striscio periferico. L’incidenza usualmente riportata è di 1 caso su individui, ma bisogna tener conto che si tratta di una malattia estremamente eterogenea da un punto di vista clinico e che molti pazienti sono completamente asintomatici. È comune soprattutto tra le popolazioni africane e mediterranee, presumibilmente perché gli ellissociti conferiscono una resistenza maggiore alla malaria. Nella maggior parte dei casi la malattia è trasmessa con meccanismo autosomico dominante.

40
Fisiopatologia Il principale difetto biochimico è rappresentato dall’alterazione delle interazioni orizzontali del citoscheletro della membrana del globulo rosso. Ciò determina: 1. la morfologia abnorme (ellittica o ovale) dei globuli rossi; 2. l’aumentata fragilità dei globuli rossi
 dei globuli rossi; 2. l’aumentata fragilità dei globuli rossi.")
41
Alterazioni biochimiche
Sono stati individuati difetti a carico di varie proteine del citoscheletro: a e b spectrina proteina 4.1 glicoforina C

42
a e b spectrina Proteina 4.1 Glicoforina C
Sono i difetti più numerosi. Questi soggetti presentano un’alterazione della tetramerizzazione della spectrina, con un aumento della quota di dimeri, incremento che correla bene con la severità del quadro clinico Proteina 4.1 Questa proteina svolge varie funzioni, tra cui quella di rafforzare il legame tra la b-spectrina e l’actina a livello dei complessi giunzionali. Il deficit parziale di questa proteina è associata con ellissocitosi asintomatica; il deficit completo determina un quadro clinico di anemia emolitica cronica. Glicoforina C I pazienti, rari, con deficit di questa glicoforina (e di quella D) presentano un quadro clinico di ellissocitosi lieve.
 presentano un quadro clinico di ellissocitosi lieve.")
43
Clinica dell’ellissocitosi ereditaria
Ellissocitosi lieve (o “comune”): è la forma clinica più comune. Si tratta di soggetti completamente asintomatici o con segni lievi di emolisi, che possono sviluppare un quadro di anemia emolitica in concomitanza di malattie come malaria o mononucleosi infettiva. Il dato fondamentale è la presenza di un numero elevato ( %) di ellissociti allo striscio periferico.
: è la forma clinica più comune. Si tratta di soggetti completamente asintomatici o con segni lievi di emolisi, che possono sviluppare un quadro di anemia emolitica in concomitanza di malattie come malaria o mononucleosi infettiva. Il dato fondamentale è la presenza di un numero elevato (30 100%) di ellissociti allo striscio periferico.")
45
Ellissocitosi con anemia emolitica cronica
Questi soggetti presentano il quadro clinico di anemia emolitica cronica, con resistenze osmotiche normali o diminuite, presenza allo striscio di ellissociti e poichilociti bizzarri. La splenectomia migliora nettamente il quadro clinico.

46
Nella maggioranza di questi soggetti è presente un’alterazione ellissogenica dell’a-spectrina, ereditata da un genitore, insieme con un polimorfismo, “in trans” dell’a-spectrina, denominato aV/41 perché interessante il dominio V dell’a-spectrina, e dovuto ad un gene mutato denominato aLely, ereditato dall’altro genitore. Questo gene mutato determina una ridotta sintesi di a-spectrina: di per sé, anche in omozigosi, esso non determina alterazioni evidenti; ma, se ereditato con il gene dell’ellissocitosi, aggrava marcatamente l’espressione della malattia.

49
Ellissocitosi ereditaria omozigote. Piropoichilocitosi ereditaria
Questi pazienti presentano un quadro di anemia emolitica cronica severa, che si manifesta già nei primi mesi di vita, e necessitante spesso di trasfusioni. Allo striscio si osservano ellissociti, poichilociti bizzarri, e microsferociti. Le resistenze osmotiche sono fortemente ridotte. La spenectomia migliora il quadro clinico. Si tratta di soggetti omozigoti per il gene dell’ellissocitosi o che presentano, come nella piropoichilocitosi ereditaria, oltre ad un gene ellissogenico, una carenza marcata di spectrina.

50
Diagnosi differenziale
Ellissociti possono essere evidenziati all’osservazione dello striscio periferico in numerose condizioni morbose: anemie megaloblastiche anemie sideropeniche talassemie mielodisplasie mielofibrosi Si tratta per lo più di condizioni morbose acquisite. Un’anamnesi accurata, l’esame clinico e pochi “tests” di laboratorio consentono di formulare agevolmente la diagnosi.
















 RELATORE DR. ADOLFO.>")






>")