Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
LAPAROSCOPICA CHIRURGIA D’URGENZA UNIVERSITA’ DEGLI STUDI DI MESSINA
FACOLTA’ DI MEDICINA E CHIRURGIA Corso di Laurea in Medicina e Chirurgia Corso di Laurea in Inf. Ped. LAPAROSCOPICA CHIRURGIA D’URGENZA PATOLOGIA CHIRURGICA IIN ETA’ PEDIATRICA - a.a. 2010/11

2
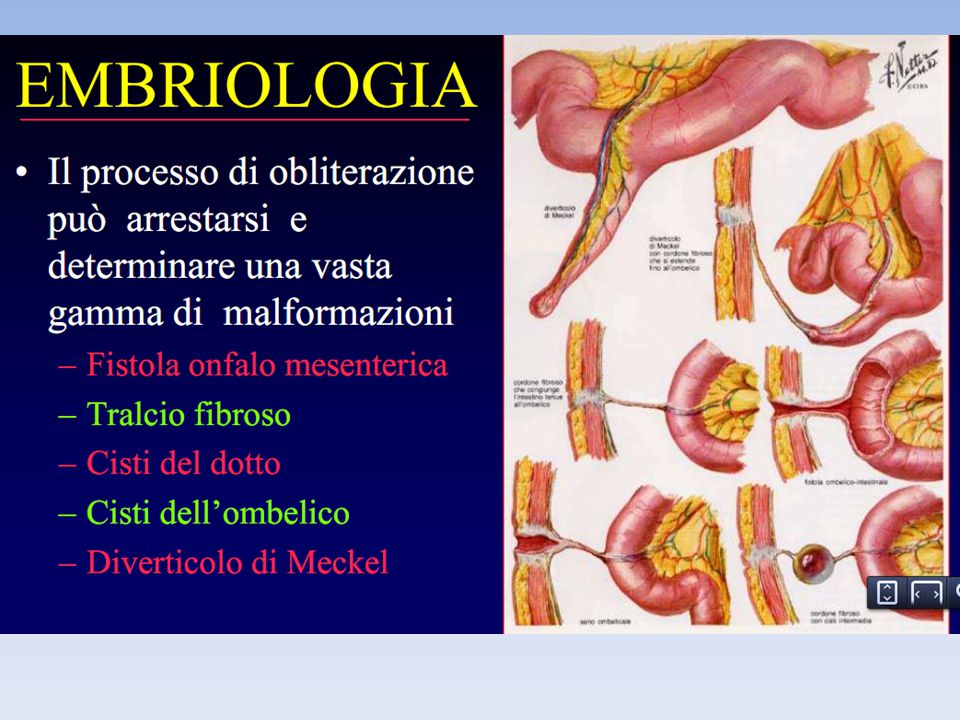
atresia congenita dell'esofago
Il termine atresia congenita dell'esofago (AE) descrive un folto gruppo di malformazioni che condividono un difetto di continuità esofagea con o senza fistola alla trachea o ai bronchi. E’ una malformazione incompatibile con la vita e la sopravvivenza e la qualità di vita dipende dalla precocità della diagnosi e da un'appropriata terapia chirurgica. La prima correzione di questa malformazione è stato eseguito da Cameron Haight presso l'Università del Michigan nel 1941.
 descrive un folto gruppo di malformazioni che condividono un difetto di continuità esofagea con o senza fistola alla trachea o ai bronchi. E’ una malformazione incompatibile con la vita e la sopravvivenza e la qualità di vita dipende dalla precocità della diagnosi e da un appropriata terapia chirurgica. La prima correzione di questa malformazione è stato eseguito da Cameron Haight presso l Università del Michigan nel")
3
preparazione preoperatoria
atresia congenita dell'esofago appropriata diagnosi preparazione preoperatoria una corretta ricostruzione della continuità esofagea con la chiusura della fistola presente nell’85% dei casi fra esofago e trachea in casi estremi: sostituzione dello stesso esofago con altro tratto di intestino. L'incidenza è di circa 1 su nati, con una lieve preponderanza per il sesso maschile (rapporto M/F di 3:2). Tra i fattori di rischio come i teratogeni ambientali: esposizione a talidomide, pillole anticoncezionali, ormoni, le malattie del sistema endocrino della madre.
. Tra i fattori di rischio come i teratogeni ambientali: esposizione a talidomide, pillole anticoncezionali, ormoni, le malattie del sistema endocrino della madre.")
5
l'anomalia può essere geneticamente determinata.
atresia congenita dell'esofago l'anomalia può essere geneticamente determinata. In primo luogo anomalie cromosomiche si verificano in 6-10% di tutti i casi compresi Trisomia 13 e 18. il rischio di ricorrenza di un secondo figlio con la stessa patologia è di circa 0,5-2,0%, mentre il rischio di un neonato nato da un genitore operato per atresia esofagea è di circa il 3,0-4,0%. La più probabile patogenesi è comunque eterogenea e multifattoriale coinvolgendo cioè diversi geni.

6
Eziologia L'embriologia del normale sviluppo dell’intestino è ancora controversa. Entro la quarta settimana di gestazione, la separazione dell'esofago dalla trachea si svolge con il ripiegamento dell’intestino primitivo. Le teorie includono malformazione del setto che divide l’intestino primitivo dalle vie aeree, fallimento di ricanalizzazione esofagea o anomalie di vascolarizzazione. Recentemente alcuni autori riportano l'ipotesi che la fistola esofago-tracheale distale nasce come un condotto cieco che termina alla biforcazione della trachea e si unisce allo stomaco. Oggi, una teoria embriologica convincente è ancora mancante. Vi è comunque una forte evidenza di una stretta relazione tra anomalie della notocorda e disturbato della segmentazione somatica risultanti in patologie vertebrali, cardiache e intestinali come l'atresia esofagea.

7
Malformazioni associate.
A causa del danno precoce nell'embriogenesi di questa malformazione è logico che bambini con atresia esofagea soffrono di un alto numero di malformazioni associate in una serie di 50-80%. Le anomalie associate più frequenti sono le malformazioni muscoloscheletriche (20-70%), seguite da quelle cardiovascolari (20-50%), genito-urinario (15-25%), gastrointestinale (15-25%) e anomalie cromosomiche (5-10%). La vasta gamma di percentuali indicate in letteratura sono determinate da differenze nell'attenzione nella diagnostica. Un attento controllo radiologico, per esempio, dell'intera colonna vertebrale e delle coste nei diversi segmenti mostrerà fino al 70% dei casi di malformazioni scheletriche associate
, seguite da quelle cardiovascolari (20-50%), genito-urinario (15-25%), gastrointestinale (15-25%) e anomalie cromosomiche (5-10%). La vasta gamma di percentuali indicate in letteratura sono determinate da differenze nell attenzione nella diagnostica. Un attento controllo radiologico, per esempio, dell intera colonna vertebrale e delle coste nei diversi segmenti mostrerà fino al 70% dei casi di malformazioni scheletriche associate.")
8
L'anomalia cardiaca più comune è quella difetto del setto ventricolare (19%), che è associata con un tasso massimo di mortalità del 16%. Altre anomalie comuni comprendono difetto del setto interatriale (20%), la tetralogia di Fallot (5%), la coartazione (1%) o l’arco aortico destro (4%). La più comune anomalia gastrointestinale associata è l'atresia ano-rettale (9%) seguita dall'atresia duodenale (5%), malrotazione (4%) e da altre atresie intestinali (1%). Ulteriori malformazioni possono coinvolgere quasi tutti i sistemi ed organi principali oltre che associarsi ad onfalocele, difetti del tubo neurale, ernia diaframmatica o altro.
 seguita dall atresia duodenale (5%), malrotazione (4%) e da altre atresie intestinali (1%). Ulteriori malformazioni possono coinvolgere quasi tutti i sistemi ed organi principali oltre che associarsi ad onfalocele, difetti del tubo neurale, ernia diaframmatica o altro.")
9
Nel 10% dei casi i pazienti affetti da atresia esofagea possono essere fatti rientrare in specifiche sindromi. Fra queste per esempio la: sindrome di Holt, di Oram, di DiGeorge, di Goldenhair, le trisomie , l'associazione CHARGE (coloboma, difetti cardiaci, atresia delle Coane, ritardo mentale, anomalie dei genitali, malformazioni dell'orecchio) e molte altre.
 e molte altre.")
10
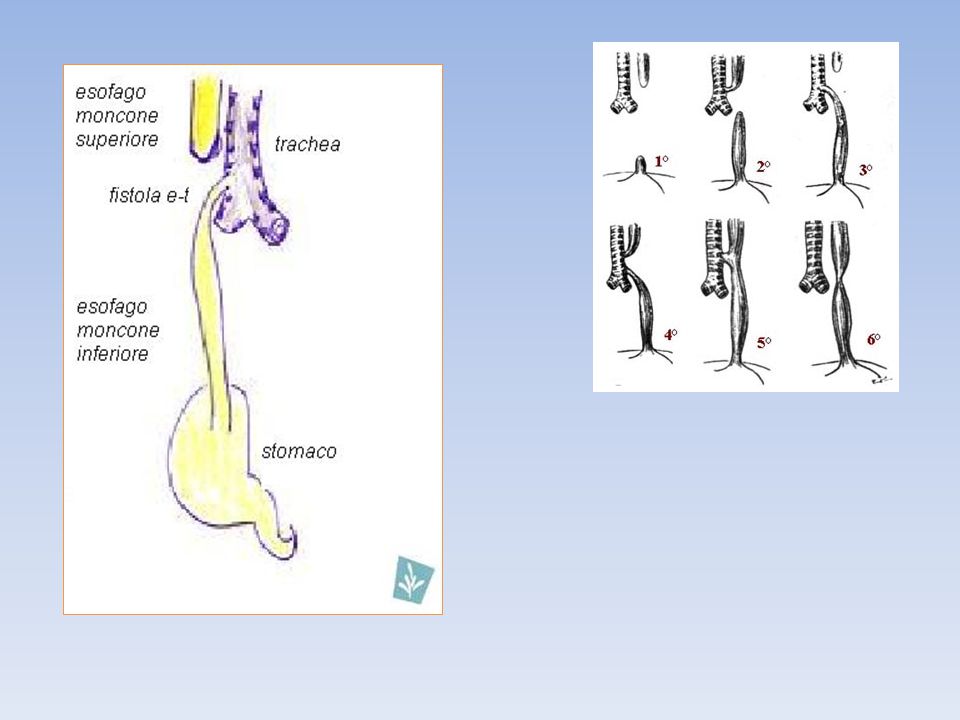
Classificazione Le varie classificazioni di solito prendono il loro orientamento dalla presenza e tipo di fistola tracheo-esofagea. Comunemente i sistemi utilizzati sono quelli descritti da Vogt (1929)e Gross (1953). Il tipo 1a di Vogt è estremamente raro ed è caratterizzato da un più o meno totale mancanza dell'esofago e non è incluso nella classica classificazione di Gross. La fistola tracheoesofagea isolata (tipo H) è classificata fra queste malformazioni anche se in realtà non vi è interruzione di continuità esofagea. Un elenco completo di tutte le variazioni nell'atresia esofagea è riassunta nel lavoro di tesi Kluth nel 1976. Classicamente si definisce: tipo 1: atresia senza fistola esofago-tracheale tipo 2: atresia con fistola esofago-tracheale al moncone superiore tipo 3: atresia con fistola esofago-tracheale al moncone inferiore (85%) tipo 4: atresia con fistola esofago-tracheale su entrambi i monconi tipo 5: solo fistola esofago-tracheale.
e Gross (1953). Il tipo 1a di Vogt è estremamente raro ed è caratterizzato da un più o meno totale mancanza dell esofago e non è incluso nella classica classificazione di Gross. La fistola tracheoesofagea isolata (tipo H) è classificata fra queste malformazioni anche se in realtà non vi è interruzione di continuità esofagea. Un elenco completo di tutte le variazioni nell atresia esofagea è riassunta nel lavoro di tesi Kluth nel Classicamente si definisce: tipo 1: atresia senza fistola esofago-tracheale. tipo 2: atresia con fistola esofago-tracheale al moncone superiore. tipo 3: atresia con fistola esofago-tracheale al moncone inferiore (85%) tipo 4: atresia con fistola esofago-tracheale su entrambi i monconi. tipo 5: solo fistola esofago-tracheale.")
11
In particolare si indicano:
Class. anatomica VOGT-types GROSS-types % Assenza dell’esofago 1 - - AE senza fistola 1b A 8.5 AE con fistola prossimale 3a B 1.0 AE con fistola distale 3b C 85.0 AE con fistola pross. e distale 3c D 1.5 Solo fistola esofago-tracheale 2 E 4.0

12
In aggiunta alla classificazione anatomica vi sono poi classificazione di rischio basate sul peso alla nascita, anomalie cardiache, patologie polmonari, che permettono di confrontare i risultati delle diverse casistiche. Le malformazioni cardiache sono quelle che hanno un maggior significato sul tasso di mortalità in questi piccoli pazienti. La classificazione più conosciuta prende il nome da Waterston (1962), tuttavia una classificazione più adeguata ai recenti progressi in medicina neonatale è quella pubblicata da Spitz nel 1994.
, tuttavia una classificazione più adeguata ai recenti progressi in medicina neonatale è quella pubblicata da Spitz nel")
13
WATERSTONE - Risk classification Type Survival (%)
A: Birthweight > g otherwise healthy 100 B: Birthweight 2.000–2.500 g or higher mild pneumonia or 85 moderate cardiac anomalies C: Birthweight < g or higher, severe pneumonia or severe 65 associated cardiac anomalies SPITZ - Risk classification Type Survival(%) I: Birthweight > g otherwise healthy 95 II: Birthweight < g or major cardiac anomaly 59 III: Birthweight < g major cardiac anomaly 22
 I: Birthweight > g otherwise healthy 95. II: Birthweight < g or major cardiac anomaly 59. III: Birthweight < g major cardiac anomaly 22.")
14
Diagnosi. Caratteristiche cliniche. Un segno indicativo di sospetta atresia dell'esofago in corso di gravidanza è il polidramnios. Polidramnios è una manifestazione aspecifica di un disturbo della deglutizione o della mancata progressione del contenuto intestinale nel feto. L'ecografia prenatale può inoltre rivelare uno spostamento in avanti ed indietro dei fluidi nella tasca esofagea superiore e, nei casi di assenza della fistola, una scarsità di fluido in stomaco e intestino tenue. Recentemente la risonanza magnetica fetale ha acquisito una maggiore attenzione per la diagnosi prenatale nelle anomalie congenite. La presentazione postnatale della malformazione è caratterizzato da drooling di saliva, soffocamento, tosse ed episodi di cianosi. Una prova di alimentazione è controindicata con questi sintomi perché è la causa prima di aspirazione e polmonite. Il successivo step diagnostico è quello di posizionare un sondino naso-gastrico. Se questo non è possibile con arresto della progressione della sonda a circa cm dall'ADS, la diagnosi di atresia esofagea è quasi certa. Per questo motivo sondini di piccolo calibro devono essere evitati in quanto arrotolandosi nella tasca superiore possono dare l'illusione di essersi spinti fin nello stomaco. L'esame obiettivo deve essere completato con la ricerca di eventuali malformazioni associate.

15
La diagnostica strumentale.
eseguire una semplice radiografia comprensiva di collo, torace e addome. Aria al di sotto del diaframma può essere vista in presenza di una fistola tracheoesofagea distale e ulteriori livelli idroaerei possono indicare un'atresia duodenale o intestinale. Un addome privo di gas indica un'atresia esofagea senza fistola al moncone inferiore. La traslucenza dei polmoni fornisce le informazioni su una possibile polmonite ab ingestis. La valutazione cardiologica comprende un'ecocardiografia di routine per valutare le malformazioni associate e la posizione dell'arco aortico, importante per valutare l'accesso chirurgico. Le altre indagini prevedono l'ecografia addominale e lo studio di eventuali anomalie schelettriche della colonna vertebrale o delle coste. L'Analisi cromosomica è eseguita solo se l'aspetto del bambino è sospetto per un difetto genetico.

16
Management preoperatorio.
I bambini sono allettati in terapia intensiva neonatale. L' intervento chirurgico immediato è raramente richiesto; di conseguenza, tutte le suddette indagini possono essere eseguite passo per passo. L'inserimento di un tubo oro- o nasoesofageo tipo Replongle doppio lume è obbligatorio per l'aspirazione continua o intermittente della saliva al fine di prevenirne l'inalazione. Il bambino deve essere posti in posizione anti-trendelemburg per ridurre al minimo il reflusso gastroesofageo nella trachea e nei polmoni attraverso la fistola inferiore. L'intubazione e la ventilazione è necessaria solo in caso di difficoltà respiratoria, polmonite grave, o altre malformazioni che compromettono la respirazione spontanea. In questi casi il tubo endotracheale deve essere possibilmente posizionato al di là della fistola distale per evitare insufflazione di gas nello stomaco. Antibiotici ad ampio spettro, la fluidoterappia e analogo della vitamina K vengono somministrati prima dell'intervento chirurgico. In presenza di una grave polmonite grave l'intervento chirurgico deve essere rinviato fino a quando il polmone non recupera. Nel caso di gravi malformazioni associate (ad esempio ernia diaframmatica o grave malformazione cardiaca), la decisione dovrebbe essere presa caso per caso.
, la decisione dovrebbe essere presa caso per caso.")
18
Gestione chirurgica dell'atresia con fistola esofagotracheale distale (85%).
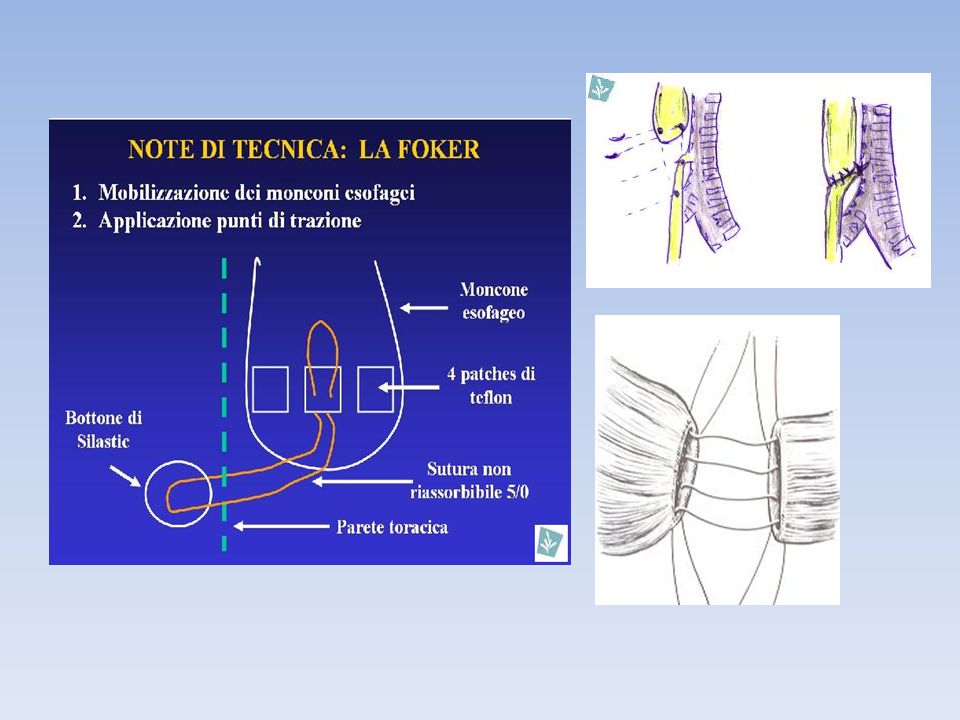
La riparazione chirurgica viene eseguita in anestesia generale con intubazione endotracheale. La fistola è in genere localizzata a circa 5-7 mm al di sopra della carena. Eccezionalmente può essere trovata alla carena o nel bronco principale destro indicando la presenza di un pouch inferiore corto e quindi gap esofageo lungo. Il passo successivo è quello di cercare una fistola del moncone superiore. La regione membranosa della parete tracheale deve essere controllata con attenzione fino alla cartilagine cricoide. Se piccola la fistola superiore può scappare alla diagnosi tracheoscopica L'obiettivo della procedura chirurgica è quello di dividere la fistola per chiuderla sul lato della trachea ed eseguire quindi un'anastomosi termino-terminale fra i due monconi esofagei. L'approccio standard è attraverso una toracotomia dorsale laterale destra per via extrapleurica attraverso il quarto spazio intercostale. Recentemente, alcuni autori hanno eseguito con successo la riparazione dell'atresia per via toracoscopica, ma questa è una procedura di elevata difficoltà e necessita pertanto di un'elevata esperienza in chirurgia endoscopica. Se vi è l'arco aortico a destra vi è indicato un approccio tramite una toracotomia sinistra.

20
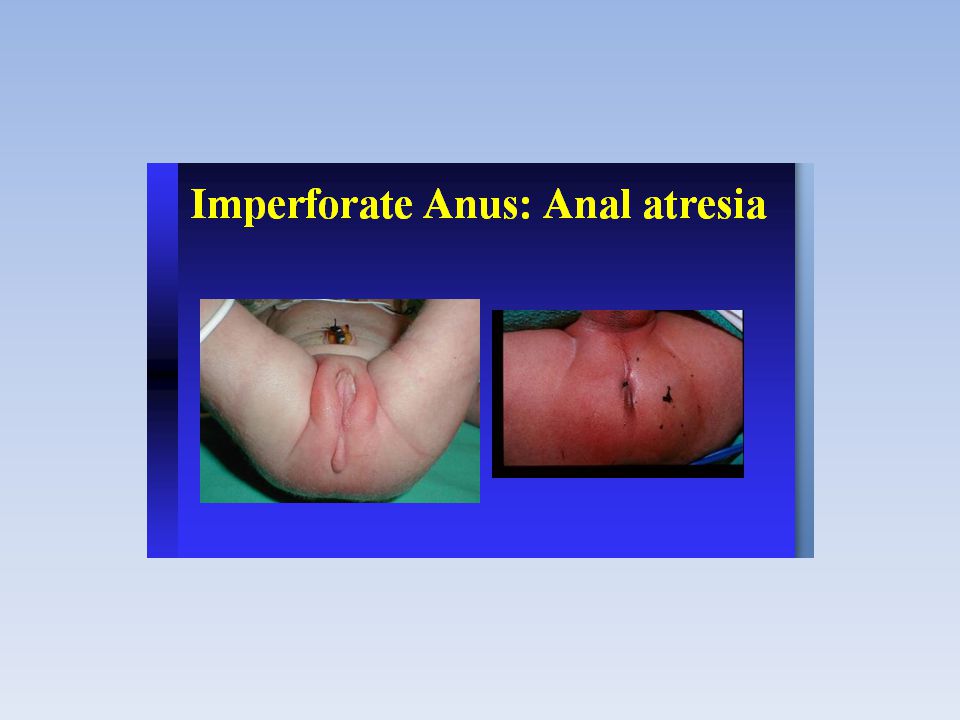
Ano imperforato

21
Ano imperforato 'Ano imperforato' è un termine impreciso utilizzato comunemente per riferirsi ad uno spettro di malformazioni anorettali, che varia da un lieve difetto che richiede un'intervento minore e presenta prognosi eccellente a malformazioni complesse con un'elevata incidenza di difetti associati che richiedono procedure chirurgiche sofisticate e specializzate. Nonostante ciò, il 30% dei pazienti nati con questi difetti presenta ancora incontinenza fecale dopo trattamento chirurgico tecnicamente corretto, ed un altro 30% presenta problemi funzionali alla defecazione quali stipsi e vari gradi incontinenza occasionale. Queste malformazioni si verificano in approssimativamente 1:4000 a 1:5000 nati. La probabilità di avere un secondo bambino colpito da questo tipo di difetto è approssimativamente dell'1% (Wyllie and Hyams. Pediatric Gastrointestinal Disease. Second Edition, 1999).
.")
22
Ano imperforato Trattasi di ano imperforato Difetto presente fin dalla nascita dove un segmento del duodeno è privo di lume Può essere anche una forma di stenosi, o più tipicamente un'ostruzione della valvola cardiaca polmonare, in tal caso è un difetto cardiaco di tipo congenito.

23
atresia biliare Sintomatologia
si intende l'assenza congenita o il mancato sviluppo di una qualche parte riguardante il sistema biliare, l'anomalia comporta immediatamente un danno all'organismo di tipo epatico. Tale forma di atresia è rara, in quanto colpisce 1:10000 nati vivi. atresia biliare Sintomatologia Fra i sintomi e i segni clinici si presentano ittero (si sviluppa intorno alla seconda o terza settimana di vita del bambino), il bambino assume in parte un colorito che tende al colore della creta, ritardo della crescita e ipertensione portale. L'anomalia dovrebbe essere tratta al più presto, per il pericolo che si sviluppi in cirrosi.
, il bambino assume in parte un colorito che tende al colore della creta, ritardo della crescita e ipertensione portale. L anomalia dovrebbe essere tratta al più presto, per il pericolo che si sviluppi in cirrosi.")
24
Patogenesi Si conoscono due forme di atresia biliare: La forma fetale, che rappresenta il 20% dei casi, ed è associata ad altre anomalie che risultano dall'inefficace stabilizzazione della lateralità del torace e dello sviluppo degli organi addominali. La forma perinatale in cui il sistema biliare normalmente formato viene distrutto dopo la nascita. Sebbene le cause esatte siano ancora sconosciute, è sospetto il coinvolgimento di infezioni virali di lunga durata, in particolare da reovirus e rotavirus Anche l'ereditarietà genetica è chiamata in causa visto i casi di atresia biliare verificatesi in gemelli e in famiglie con anomalie del sistema biliare intraepatico.

25
Morfologia Le caratteristiche salienti comprendono infiammazione e stenosi fibrosante del dotto epatico o comune, infiammazione periduttale dei dotti biliari intraepatici e progressiva distruzione del sistema biliare intraepatico. Diagnosi differenziale Se nello sviluppo del bambino nei primi giorni si formasse la semplice epatite neonatale si potrebbe curare senza il trapianto, mentre al contrario la cirrosi è quasi sempre mortale, per questo si rende molto importante una corretta diagnosi. Terapia Fra i vari trattamenti possibili quello chirurgico spesso non risulta essere definitivo, in tali casi occorre il trapianto del fegato.

26
Ano imperforato Eziologia Le cause dell'ostruzione non sono a tutt'oggi state definite e molte cause diverse fra loro sono ritenute possibili. Il trattamento deve essere chirurgico e precoce. Una diagnosi effettuata entro il secondo mese di vita del bambino, aumenta di molto le possibilità di una prognosi positiva. È fisiologico, nei neonati, la presenza di un ittero neonatale, che però non si protrae di norma oltre i 15 giorni al massimo dalla nascita. Il perdurare o l'insorgere di un ittero dopo i quindici giorni deve indurre, rapidamente, ad analisi più approfondite. La rarità della patologia fa sì che essa possa essere di fatto sconosciuta a molti pediatri.

27
La terapia chirurgica La mortalità della malattia era in pratica del 100% fino all'introduzione, alla fine degli anni '50, della tecnica chirurgica detta di Kasai, dal nome del chirurgo giapponese che per primo la mise in opera, o anche portoenterostomia. La diagnosi precoce è uno dei principali fattori di successo di questa tecnica chirurgica. Attualmente, le possibilità di sopravvivenza dei bambini nati con questa patologia sono drasticamente migliori, sia con il solo trattamento chirurgico che, nel caso di insuccesso di questo, con la possibilità di ricorrere ad un trapianto di fegato.

28
atresia biliare extraepatica
L'atresia biliare extraepatica è una grave e rara malattia neonatale, che interessa il fegato e le vie biliari. La sua incidenza è di circa 1 bambino su nati. Nei bambini colpiti da questa patologia si ha una ostruzione dei dotti biliari, con conseguente riversamento della bile nel circolo sanguigno, che provoca ittero, urine colorate e presenza difeci acoliche, vale a dire di colore molto chiaro (proprio a causa della mancanza della bile). Uno dei fattori che determinano la possibilità di successo dell'intervento chirurgico è il livello di ostruzione delle vie biliari.
. Uno dei fattori che determinano la possibilità di successo dell intervento chirurgico è il livello di ostruzione delle vie biliari.")
29
Atresia polmonare Atresia polmonare Può essere anche una forma di stenosi, o più tipicamente un'ostruzione della valvola cardiaca polmonare, in tal caso è un difetto cardiaco di tipo congenito, ad esempio la tetralogia di Fallot.

30
La tetralogia di Fallot (detta anche sindrome del bambino blu) è una malformazione cardiaca congenita che classicamente ha quattro elementi anatomici. Fu descritta per la prima volta nel 1672 da Niccolò Stenone. Nel 1888 il medico francese Étienne-Louis Arthur Fallot ha descritto esattamente le quattro caratteristiche anatomiche: La comunicazione fra i due ventricoli, le due parti pompanti del cuore (difetto interventricolare). L'origine biventricolare dell'aorta, che si trova a cavallo fra i due ventricoli, sopra il difetto interventricolare (Aorta a cavaliere). Una stenosi (restringimento) sottovalvolare e valvolare polmonare. Un'ipertrofia (cioè ingrossamento muscolare) del ventricolo destro, come conseguenza degli altri difetti. tetralogia di Fallot
. L origine biventricolare dell aorta, che si trova a cavallo fra i due ventricoli, sopra il difetto interventricolare (Aorta a cavaliere). Una stenosi (restringimento) sottovalvolare e valvolare polmonare. Un ipertrofia (cioè ingrossamento muscolare) del ventricolo destro, come conseguenza degli altri difetti. tetralogia di Fallot.")
31
Questa patologia nasce perché, già in fase embriologica, la parte superiore e quella inferiore del cuore non combaciano provocando una ostruzione all’efflusso verso l’arteria polmonare con le conseguenze sopra indicate. Il bambino appare blu, cioè con cianosi periferiche e perilabiale. Le dita a bacchetta di tamburo si formano per il miscelamento di sangue (sangue non completamente ossigenato) dovuto al difetto interventricolare. La cianosi può essere: centrale: quando di origine polmonare o cardiaca, in caso per esempio di tetralogia di Fallot periferica: in caso di aumentata richiesta di sangue da parte dei tessuti.

32
atresia vaginale atresia vaginale è un'anomalia congenita dove si mostra una deformazione funzionale della vagina. Eziologia Deriva dalla mancata canalizzazione del tratto terminale di dotti di Muller, da cui deriva l'organo. Si manifesta generalmente alla pubertà poiché il sangue mestruale non riuscendo a defluire all'esterno, determina ematometro (sangue che si raccoglie in utero), ematosalpinge (sangue nelle tube), causando coliche addominali.
, ematosalpinge (sangue nelle tube), causando coliche addominali.")
33
imene cribroso oppure addirittura essere imperforato.
L'imene è una membrana epitelio-connettivale elastica che precede l'introito vaginale. La sua conformazione è in genere anulare, ma può presentare un setto mediano o dei microfori (imene cribroso) oppure addirittura essere imperforato. Nell' ultimo caso è presente alla prima mestruazione un ristagno di sangue e tessuto endometriale chiamato ematocolpo che si trasforma in ematometra(ristagno di sangue nell'utero): tale situazione è di estrema gravità e necessita di un tempestivo intervento del Ginecologo. E' ovvio quindi che la tua situazione anatomica, almeno in questo senso, sia del tutto normale. E poi vero che il tessuto che compone l'imene può essere molto elastico (imene compiacente) permettendo una prima penetrazione senza lacerazione e senza alcuna perdita di sangue, oppure molto rigido. In tale caso, alcune volte, e necessario un piccolissimo intervento che potra essere eseguito anche in anestesa loco.reginale.
 oppure addirittura essere imperforato. Nell ultimo caso è presente alla prima mestruazione un ristagno di sangue e tessuto endometriale chiamato ematocolpo che si trasforma in ematometra(ristagno di sangue nell utero): tale situazione è di estrema gravità e necessita di un tempestivo intervento del Ginecologo. E ovvio quindi che la tua situazione anatomica, almeno in questo senso, sia del tutto normale. E poi vero che il tessuto che compone l imene può essere molto elastico (imene compiacente) permettendo una prima penetrazione senza lacerazione e senza alcuna perdita di sangue, oppure molto rigido. In tale caso, alcune volte, e necessario un piccolissimo intervento che potra essere eseguito anche in anestesa loco.reginale.")
34
atresia digiunoileale
atresia digiunoileale in campo medico, si intende una malformazione congenita del nascituro del digiuno, che non è stato completato Tipologia Esistono 5 tipi diversi di atresia digiunoileale a seconda dell'interessamente dell'intestino: Tipo I, intestino indenne Tipo II, Tipo III a, Tipo III b, Tipo IV, l'atresia coinvolge molteplici segmenti dell'intestino. La radiografia dell'addome è l'esame diagnostico, anche per comprendere di quale tipologia si tratta.

35
Sintomatologia I sintomi si manifestano rapidamente, già nei primi giorni di vita del bambino, distensione addominale e difficoltà nel defecare. Eziologia La causa è conseguente ad un'ischemia [modifica]Terapia Il trattamento chirurgico risolve la quasi totalità dei casi, prima di effettuarla si sospende la normale nutrizione del soggetto sostituendola con quella endovena di liquidi. [modifica]Prognosi L'andamento futuro della persona è strettamente rilegato alla lunghezza della parte dell'intestino tenue rimanente. Può comportare la nascita della sindrome dell'intestino corto.

36
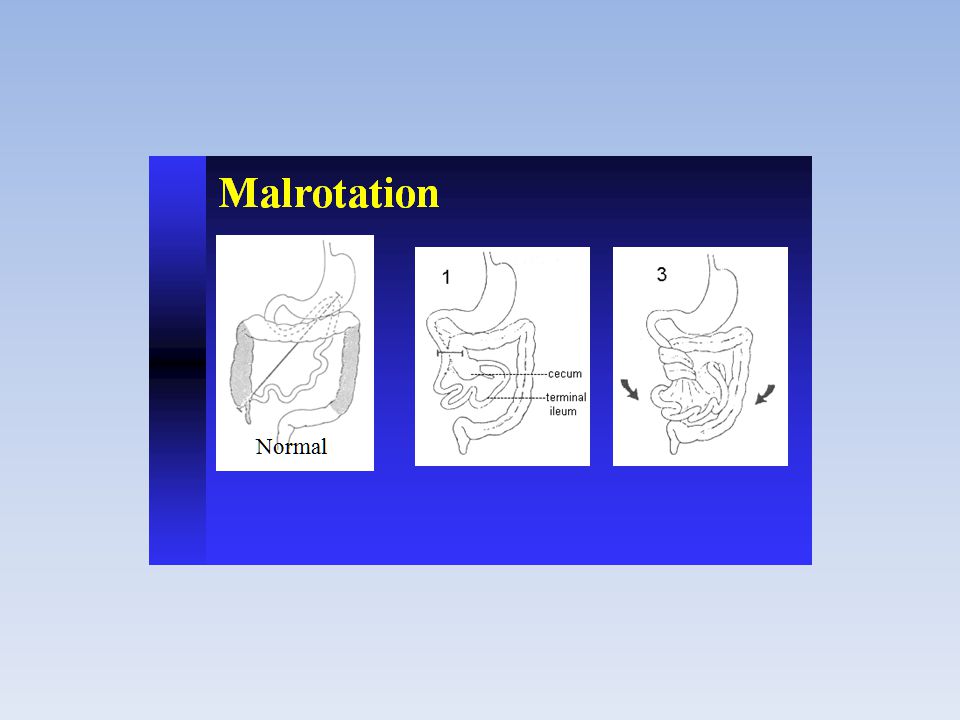
atresia duodenale atresia duodenale in campo medico, si intende una malformazione congenita dei nascituri, una forma di atresia gastrointestinale Epidemiologia Per quanto riguarda le varie forme di atresia possibili, quella duodenale è una delle più frequenti nei nascituri, arrivando come incidenza da 1 su ad 1 su a seconda degli studi effettuati[1] Il 25% di tutti i bambini affetti da tale malformazione sono soggetti alla sindrome di Down Eziologia La causa è una mancata canalizzazione durante la fase embrionale del duodeno. Sintomatologia Fra i sintomi e i segni clinici ritroviamo polidramnios, vomito e sovente si assiste a malrotazione intestinale Esami Per una corretta diagnosi è sufficiente la radiografia, dove si mostra un segno distintivo della patologia, una doppia bolla, una nel duodeno e l'altra nello stomaco.

37
atresia duodenale duplicazione intestinale, in campo medico, si intende una particolare struttura anomala, rivestita da epitelio mucoso, costituita dalla formazione di cisti o piccoli canali che si attaccano al tessuto interno dell'intestino. Tale difformità si riscontra durante lo sviluppo del feto ed è causata da un difetto del processo di vacuolizzazione.

38
stenosi duodenale stenosi duodenale è una forma di stenosi (restringimento) tipica dell'apparato gastrointestinale, ed è una complicanza della sindrome di Down
 tipica dell apparato gastrointestinale, ed è una complicanza della sindrome di Down.")
39
duplicazione intestinale, in campo medico, si intende una particolare struttura anomala, rivestita da epitelio mucoso, costituita dalla formazione di cisti o piccoli canali che si attaccano al tessuto interno dell'intestino. Tale difformità si riscontra durante lo sviluppo del feto ed è causata da un difetto del processo di vacuolizzazione. duplicazione intestinale Epidemiologia La sua frequenza nei nascituri è rara, mostrandosi quasi esclusivamente nei primi anni del bambino. I luoghi dove si manifestano con più frequenza sono l'ileo, l'esofago e lo stomaco, e con minore incidenza colon e duodeno. Sintomatologia Una persona può essere affetta da più duplicazioni intestinali e non accorgesene, oppure possono mostrarsi sintomi a livello gastrointestinale con dolore persistente. Terapia Si interviene solo per le duplicazioni che mostrano sintomi gravi, il trattamento è chirurgico con resezione completa delle malformazioni anatomiche.

40
atresia della tricuspide
atresia della tricuspide in campo medico, si intende una malattia cardiaca (cardiopatia) , la cui caratteristica principal duplicazione intestinale e è la presenza di un'ostruzione di una qualche natura del flusso sanguigno che passa da l'atrio destro al ventricolo destro, la cui causa è il malfunzionamento o addirittura l'assenza della valvola tricuspide. Tale difetto si accompagna solitamente con altri, sempre congeniti.
 , la cui caratteristica principal duplicazione intestinale e è la presenza di un ostruzione di una qualche natura del flusso sanguigno che passa da l atrio destro al ventricolo destro, la cui causa è il malfunzionamento o addirittura l assenza della valvola tricuspide. Tale difetto si accompagna solitamente con altri, sempre congeniti.")
41
Il tumore di Wilms (TW) o nefroblastoma rappresenta a tutt'oggi un esempio dei progressi raggiunti nella cura delle neoplasie pediatriche. L'elaborazione di approcci multidisciplinari con chemio-radioterapia pre e/o post-chirurgica nell'ambito di studi cooperativi ha permesso di migliorarne le possibilità di cura, ottenendo sopravvivenze dell'80-90%. tumore di Wilms EPIDEMIOLOGIA Il TW nel 75% dei casi insorge prima dei 5 anni d'età, con un'incidenza annua di 7 casi/milione. Nel 5% dei bambini può essere bilaterale. Malformazioni congenite isolate (quali emipertrofia, aniridia, anomalie genito-urinarie) o raggruppabili in sindromi definite (Beckwith-Wiedeman, Denys-Drash, WAGR) possono associarsi al nefroblastoma. EZIOLOGIA I fattori eziologici non sono noti, tuttavia l'associazione tra TW e sindromi malformative suggerisce l'importanza di fattori genetici nell'eziopatogenesi del nefroblastoma. Mutazioni del gene oncosoppressore WT1, situato sul braccio corto del cromosoma 11 (11p13), sono presenti nel 10% dei casi testati. ISTOPATOLOGIA Sono stati identificati quadri istologici correlati ad una prognosi meno favorevole, caratterizzati dalla presenza di anaplasia, nonché forme di neoplasia renale ora distinte dal nefroblastoma, quali il sarcoma a cellule chiare e il tumore rabdoide. QUADRO CLINICO La presentazione clinica prevalente è la massa addominale con dolore e incremento volumetrico dell'addome. Altri possibili segni sono ematuria macroscopica ed ipertensione arteriosa. INDAGINI DIAGNOSTICHE L'ecografica dell'addome, completata dalla TAC di torace e addome, costituiscono le indagini radiologiche elettive per l'orientamento diagnostico e per una valutazione prechirurgica. Gli esami di laboratorio sono quelli di routine.
 o raggruppabili in sindromi definite (Beckwith-Wiedeman, Denys-Drash, WAGR) possono associarsi al nefroblastoma. EZIOLOGIA I fattori eziologici non sono noti, tuttavia l associazione tra TW e sindromi malformative suggerisce l importanza di fattori genetici nell eziopatogenesi del nefroblastoma. Mutazioni del gene oncosoppressore WT1, situato sul braccio corto del cromosoma 11 (11p13), sono presenti nel 10% dei casi testati. ISTOPATOLOGIA Sono stati identificati quadri istologici correlati ad una prognosi meno favorevole, caratterizzati dalla presenza di anaplasia, nonché forme di neoplasia renale ora distinte dal nefroblastoma, quali il sarcoma a cellule chiare e il tumore rabdoide. QUADRO CLINICO La presentazione clinica prevalente è la massa addominale con dolore e incremento volumetrico dell addome. Altri possibili segni sono ematuria macroscopica ed ipertensione arteriosa. INDAGINI DIAGNOSTICHE L ecografica dell addome, completata dalla TAC di torace e addome, costituiscono le indagini radiologiche elettive per l orientamento diagnostico e per una valutazione prechirurgica. Gli esami di laboratorio sono quelli di routine.")
42
STORIA NATURALE La neoplasia, generalmente confinata all'organo di origine, può talvolta superarne i limiti anatomici e infiltrare i tessuti circostanti. Metastasi a distanza possono essere evidenti già alla diagnosi nel 10% dei casi, coinvolgendo principalmente i polmoni. STRATEGIA TERAPEUTICA L'intervento chirurgico di nefrectomia con eventuale dissezione dei linfonodi regionali e para-aortici omolaterali al tumore costituisce il momento terapeutico più importante. Una chirurgia conservativa del rene è indicata solamente nelle situazioni di bilateralità della neoplasia. Il tempo ideale in cui effettuare la nefrectomia è a tutt'oggi oggetto di dibattito. I ricercatori nordamericani ed il gruppo italiano A.I.E.O.P. prediligono la chirurgia come primo atto terapeutico e successivamente, in base alla "reale" stadiazione chirurgica, la chemioterapia complementare. Il gruppo europeo della SIOP (Società Internazionale di Oncologia Pediatrica) ha invece condotto una serie di studi clinici per testare l'efficacia della chemioterapia pre-operatoria, che consente un approccio chirurgico più agevole. I risultati terapeutici ottenuti con queste due strategie sono di fatto sovrapponibili. Il trattamento farmacologico riveste comunque un ruolo importante. I farmaci più attivi sono actinomicina-D, vincristina e adriamicina. Nei pazienti non responsivi a questi farmaci, ifosfamide e VP16 si sono dimostrati efficaci nell'indurre la remissione. Il ruolo della radioterapia è sottoposto a rivalutazione critica, trovando indicazione solo in casi con malattia più avanzata. Le conclusioni cui sono giunti gli studi multicentrici nordamericani per quanto riguarda la chemioterapia complementare alla chirurgia sono le seguenti: nei bambini con stadio I e II ed istologia favorevole l'associazione vincristina + actinomicina D costituisce il trattamento di scelta, per periodi di 2-6 mesi in relazione allo stadio; nei pazienti classificati allo stadio III l'aggiunta di adriamicina e della radioterapia ha permesso di migliorare la sopravvivenza. Situazioni terapeutiche particolari, che richiedono trattamenti più aggressivi o personalizzati sono rappresentate rispettivamente dal nefroblastoma al IV (metastasi a distanza) o V stadio (neoplasie bilaterali) e dai tumori con istologia sfavorevole. Non esistono attualmente dati conclusivi circa il vantaggio di una chemioterapia ad alte dosi rispetto alla chemioterapia a dosaggi convenzionali in quest'ultimo gruppo di pazienti
 ha invece condotto una serie di studi clinici per testare l efficacia della chemioterapia pre-operatoria, che consente un approccio chirurgico più agevole. I risultati terapeutici ottenuti con queste due strategie sono di fatto sovrapponibili. Il trattamento farmacologico riveste comunque un ruolo importante. I farmaci più attivi sono actinomicina-D, vincristina e adriamicina. Nei pazienti non responsivi a questi farmaci, ifosfamide e VP16 si sono dimostrati efficaci nell indurre la remissione. Il ruolo della radioterapia è sottoposto a rivalutazione critica, trovando indicazione solo in casi con malattia più avanzata. Le conclusioni cui sono giunti gli studi multicentrici nordamericani per quanto riguarda la chemioterapia complementare alla chirurgia sono le seguenti: nei bambini con stadio I e II ed istologia favorevole l associazione vincristina + actinomicina D costituisce il trattamento di scelta, per periodi di 2-6 mesi in relazione allo stadio; nei pazienti classificati allo stadio III l aggiunta di adriamicina e della radioterapia ha permesso di migliorare la sopravvivenza. Situazioni terapeutiche particolari, che richiedono trattamenti più aggressivi o personalizzati sono rappresentate rispettivamente dal nefroblastoma al IV (metastasi a distanza) o V stadio (neoplasie bilaterali) e dai tumori con istologia sfavorevole. Non esistono attualmente dati conclusivi circa il vantaggio di una chemioterapia ad alte dosi rispetto alla chemioterapia a dosaggi convenzionali in quest ultimo gruppo di pazienti.")
43
IL MELANOMA DELLA CUTE IN ETA’ PEDIATRICA
Aspetti clinici Il melanoma cutaneo è un tumore maligno ad origine dai melanociti ed è una neoplasia tipica dell’età adulta (5% di tutti i tumori), la cui incidenza è in costante aumento, probabilmente correlata all’esposizione ai raggi ultravioletti. In età pediatrica, però, il melanoma è un tumore molto raro: solo il 2% di tutti i melanomi insorge in pazienti di età inferiore a 20 anni, la maggior parte dei quali sono adolescenti. Come conseguenza, l’esperienza clinica nei bambini è scarsa e la gestione clinica viene mutuata dalle esperienze sui pazienti adulti. Il momento fondamentale nella storia clinica del melanoma è quello del riconoscimento diagnostico ed ogni sforzo deve essere fatto per facilitare una diagnosi precoce. Esistono criteri clinici per identificare un lesione pigmentata (nevo) come “a rischio”: la regola mnemonica ABCDE suggerisce che un nevo è sospetto se subisce delle modificazioni in forma (Asimmetria), margini (Bordi irregolari), pigmentazione (Colore scuro e/o variegato), diametro (Dimensioni superiori a 5 mm) e se appare rapidamente evolutivo (Evoluzione). I criteri utilizzati nell’adulto possono in realtà valere meno nel bambino in fase di crescita, dove modificazioni lente e costanti di nevi preesistenti possono essere del tutto fisiologiche. Inoltre, i melanomi pediatrici spesso si presentano con aspetti atipici, cioè depigmentati, o amelanotico, e rilevati, con aspetti che possono richiamare il granuloma piogenico.
, la cui incidenza è in costante aumento, probabilmente correlata all’esposizione ai raggi ultravioletti. In età pediatrica, però, il melanoma è un tumore molto raro: solo il 2% di tutti i melanomi insorge in pazienti di età inferiore a 20 anni, la maggior parte dei quali sono adolescenti. Come conseguenza, l’esperienza clinica nei bambini è scarsa e la gestione clinica viene mutuata dalle esperienze sui pazienti adulti. Il momento fondamentale nella storia clinica del melanoma è quello del riconoscimento diagnostico ed ogni sforzo deve essere fatto per facilitare una diagnosi precoce. Esistono criteri clinici per identificare un lesione pigmentata (nevo) come a rischio : la regola mnemonica ABCDE suggerisce che un nevo è sospetto se subisce delle modificazioni in forma (Asimmetria), margini (Bordi irregolari), pigmentazione (Colore scuro e/o variegato), diametro (Dimensioni superiori a 5 mm) e se appare rapidamente evolutivo (Evoluzione). I criteri utilizzati nell’adulto possono in realtà valere meno nel bambino in fase di crescita, dove modificazioni lente e costanti di nevi preesistenti possono essere del tutto fisiologiche. Inoltre, i melanomi pediatrici spesso si presentano con aspetti atipici, cioè depigmentati, o amelanotico, e rilevati, con aspetti che possono richiamare il granuloma piogenico.")
44
Stadiazione e fattori prognostici
La sopravvivenza globale dei pazienti affetti da melanoma è stimata essere intorno al 50-60% a 10 anni, anche se negli ultimi anni la prognosi appare in continuo miglioramento in relazione all’incremento dei casi diagnosticati precocemente, con spessore inferiore al millimetro. La prognosi è strettamente correlata allo spessore (guarisce più del 90% dei casi di melanoma “sottile”). Oltre al già citato stadio patologico, correlato ai millimetri e al livello di invasione delle cellule tumorali in profondità nel derma, esiste uno stadio clinico. Il recentemente rivisto sistema di stadiazione dell’American Joint Committee on Cancer (AJCC) considera spessore del tumore (T), tipo e numero di metastasi linfonodali (N), metastasi a distanza (M). Data la tendenza del melanoma a dare metastasi linfonodali (e il peso prognostico di tale evento) è assai importante poter identificare quei soggetti che in assenza di segni clinici presentano localizzazioni occulte a livello dei linfonodi e necessitano pertanto di dissezioni linfonodali selettive. La tecnica del “linfonodo-sentinella”, basata sul presupposto biologico che la diffusione linfatica del melanoma sia sequenziale, senza salti di stazioni linfonodali, si effettua inoculando un colorante vitale e/o un isotopo radioattivo alla periferia della lesione (o della cicatrice chirurgica), identificando così il linfonodo di primo drenaggio (detto appunto linfonodo “sentinella”) e permettendo al chirurgo di effettuare una biopsia mirata. In caso di esame istologico positivo, è necessario procedere alla dissezione linfonodale.
. Oltre al già citato stadio patologico, correlato ai millimetri e al livello di invasione delle cellule tumorali in profondità nel derma, esiste uno stadio clinico. Il recentemente rivisto sistema di stadiazione dell’American Joint Committee on Cancer (AJCC) considera spessore del tumore (T), tipo e numero di metastasi linfonodali (N), metastasi a distanza (M). Data la tendenza del melanoma a dare metastasi linfonodali (e il peso prognostico di tale evento) è assai importante poter identificare quei soggetti che in assenza di segni clinici presentano localizzazioni occulte a livello dei linfonodi e necessitano pertanto di dissezioni linfonodali selettive. La tecnica del linfonodo-sentinella , basata sul presupposto biologico che la diffusione linfatica del melanoma sia sequenziale, senza salti di stazioni linfonodali, si effettua inoculando un colorante vitale e/o un isotopo radioattivo alla periferia della lesione (o della cicatrice chirurgica), identificando così il linfonodo di primo drenaggio (detto appunto linfonodo sentinella ) e permettendo al chirurgo di effettuare una biopsia mirata. In caso di esame istologico positivo, è necessario procedere alla dissezione linfonodale.")
45
Terapia La chirurgia resta la principale, se non l’unica, opzione terapeutica nel melanoma. Nel bambino, l’escissione diagnostica di una lesione pigmentata è indicata solo in caso di fondato sospetto di malignità (rapida evoluzione, morfologia chiaramente atipica); in casi dubbi, può essere suggerita la stretta osservazione clinica. Quando l’exeresi è giustificata, essa dovrebbe essere effettuata in ambito specialistico: l’ampiezza della stessa deve essere limitata per ragioni funzionali ed estetiche: un melanoma confermato istologicamente può essere radicalizzato successivamente. La terapia resta chirurgica anche in caso di malattia loco-regionale (svuotamento lnfonodale), mentre è ancora discusso il ruolo dell’immunoterapia adiuvante con -IFN ad alte dosi. La chemioterapia, in caso di malattia avanzata, ha dato negli anni risultati poco soddisfacenti, anche se recentemente l’utilizzo di nuove molecole e l’associazione con l’immunoterapia sembra aver incrementato il tasso di risposta.
; in casi dubbi, può essere suggerita la stretta osservazione clinica. Quando l’exeresi è giustificata, essa dovrebbe essere effettuata in ambito specialistico: l’ampiezza della stessa deve essere limitata per ragioni funzionali ed estetiche: un melanoma confermato istologicamente può essere radicalizzato successivamente. La terapia resta chirurgica anche in caso di malattia loco-regionale (svuotamento lnfonodale), mentre è ancora discusso il ruolo dell’immunoterapia adiuvante con -IFN ad alte dosi. La chemioterapia, in caso di malattia avanzata, ha dato negli anni risultati poco soddisfacenti, anche se recentemente l’utilizzo di nuove molecole e l’associazione con l’immunoterapia sembra aver incrementato il tasso di risposta.")
46
CARCINOMA DI DERIVAZIONE DELL’EPITELIO FOLLICOLARE DELLA TIROIDE IN ETÀ PEDIATRICA
Epidemiologia. I tumori endocrini in età pediatrica rappresentano approssimativamente il 4-5% di tutte le neoplasie osservate. Il 40-45% di questi tumori originano dalle gonadi, il 30% dalla ghiandola tiroidea ed il 20% dall’ipofisi; i rimanenti casi originano dalle paratiroidi, dalla corteccia e dalla midollare del surrene. Il termine ‘carcinoma differenziato della tiroide’ (CDT) comprende, in virtù della revisione istologica e delle caratteristiche prognostiche comuni, i seguenti istotipi di derivazione dall’epitelio follicolare: il carcinoma papillare ben della testa e del collo in età pediatrica. L’incidenza annua stimata è intornodifferenziato e sue varianti, il carcinoma follicolare capsulato (minimamente invasivo), ed i carcinomi poco differenziati. Il CDT non è comune in età pediatrica. Esso rappresenta l’1,4-3% di tutti i carcinomi diagnosticati nei bambini. Questa percentuale corrisponde a circa il 7% dei tumori allo 0,2-0,4 per milione di bambini.
 comprende, in virtù della revisione istologica e delle caratteristiche prognostiche comuni, i seguenti istotipi di derivazione dall’epitelio follicolare: il carcinoma papillare ben della testa e del collo in età pediatrica. L’incidenza annua stimata è intornodifferenziato e sue varianti, il carcinoma follicolare capsulato (minimamente invasivo), ed i carcinomi poco differenziati. Il CDT non è comune in età pediatrica. Esso rappresenta l’1,4-3% di tutti i carcinomi diagnosticati nei bambini. Questa percentuale corrisponde a circa il 7% dei tumori allo 0,2-0,4 per milione di bambini.")
47
Il CDT in età pediatrica mostra una invasività elevata sia locale che a distanza, maggiore rispetto ai casi degli adulti. Al momento della diagnosi, infatti, presenta solitamente una elevata tendenza alla invasione extratiroidea nei tessuti molli del collo, una percentuale elevata di metastasi linfonodali cervicali e di metastasi a distanza (polmone e osso). La presenza di microfocolai neoplastici multipli sia nel lobo omolaterale che controlaterale rispetto alla massa neoplastica principale è quasi la regola. La percentuale di casi con invasione vascolare (circa 30%) è molto elevata. Anche le riprese evolutive di malattia sono più comuni nei bambini rispetto agli adulti. Nonostante tutto questo, la prognosi è eccellente e la mortalità estremamente bassa, nettamente inferiore rispetto a quella degli adulti a parità di diffusione di malattia. Il motivo di questo differente comportamento biologico rimane per ora sconosciuto, ma potrebbero essere coinvolti fattori genetico-molecolari.
. La presenza di microfocolai neoplastici multipli sia nel lobo omolaterale che controlaterale rispetto alla massa neoplastica principale è quasi la regola. La percentuale di casi con invasione vascolare (circa 30%) è molto elevata. Anche le riprese evolutive di malattia sono più comuni nei bambini rispetto agli adulti. Nonostante tutto questo, la prognosi è eccellente e la mortalità estremamente bassa, nettamente inferiore rispetto a quella degli adulti a parità di diffusione di malattia. Il motivo di questo differente comportamento biologico rimane per ora sconosciuto, ma potrebbero essere coinvolti fattori genetico-molecolari..")
48
CARCINOIDE DELL’APPENDICE
Aspetti epidemiologici e clinici Il primo carcinoide dell’appendice (CA) è stato descritto nel 1882 ma il termine “karzinoid” è nato solo nel 1907 per descrivere un tipo particolare di tumore dell’intestino a comportamento più indolente rispetto al tipico adenocarcinoma intestinale. I CA sono stati descritti più frequentemente nei bronchi e nel tratto gastro-enterico, ma possono ritrovarsi anche nel pancreas, nel timo e nelle ovaie. Nonostante in età pediatrica siano molto rari, i CA dell’appendice sono comunque i tumori gastro-intestinali epiteliali più frequenti e rappresentano di regola un riscontro occasionale all’esame istologico dopo un intervento per appendicite: l’incidenza è di circa 2-5 casi ogni 1000 appendicectomie. La sindrome da carcinoide, sebbene descritta in associazione al CA dell’adulto, non è stata descritta sinora nelle casistiche pediatriche. Tale sindrome, caratterizzata da flushing, affanno respiratorio, ipertensione, diarrea, è causata da sostanze vasoattive prodotte dalle cellule tumorali: tra queste, la più conosciuta è la serotonina, sintetizzata da un precursore, il 5-idrossi triptofano, e successivamente metabolizzata in acido 5-idrossi indolacetico, che viene escreto nelle urine. In alcuni pazienti pediatrici sono stati descritti sintomi quali ipertensione e diarrea, ma in nessun caso sono stati riscontrati valori umorali elevati di serotonina o dei suoi metaboliti.
 è stato descritto nel 1882 ma il termine karzinoid è nato solo nel 1907 per descrivere un tipo particolare di tumore dell’intestino a comportamento più indolente rispetto al tipico adenocarcinoma intestinale. I CA sono stati descritti più frequentemente nei bronchi e nel tratto gastro-enterico, ma possono ritrovarsi anche nel pancreas, nel timo e nelle ovaie. Nonostante in età pediatrica siano molto rari, i CA dell’appendice sono comunque i tumori gastro-intestinali epiteliali più frequenti e rappresentano di regola un riscontro occasionale all’esame istologico dopo un intervento per appendicite: l’incidenza è di circa 2-5 casi ogni 1000 appendicectomie. La sindrome da carcinoide, sebbene descritta in associazione al CA dell’adulto, non è stata descritta sinora nelle casistiche pediatriche. Tale sindrome, caratterizzata da flushing, affanno respiratorio, ipertensione, diarrea, è causata da sostanze vasoattive prodotte dalle cellule tumorali: tra queste, la più conosciuta è la serotonina, sintetizzata da un precursore, il 5-idrossi triptofano, e successivamente metabolizzata in acido 5-idrossi indolacetico, che viene escreto nelle urine. In alcuni pazienti pediatrici sono stati descritti sintomi quali ipertensione e diarrea, ma in nessun caso sono stati riscontrati valori umorali elevati di serotonina o dei suoi metaboliti.")
49
Valutazione clinico-strumentale (successiva al riscontro)
La diagnostica successiva al riscontro di un CA è mirata alla ricerca di residui intraddominali e metastasi a distanza e si basa su una valutazione bioumorale e su quella strumentale. La letteratura è concorde sulla necessità di determinare i valori serici di serotonina e cromogranina A, una glicoproteina contenuta nella frazione solubile dei granuli neurosecretori. L’acido 5-idrossi-indolacetico (5-HIAA), metabolita della serotonina, deve essere dosato nelle urine delle 24 ore. Poiché la maggior parte dei C esprime recettori per la somatostatina, l’indagine di scelta per l’individuazione di lesioni residue extraddominali ed extraepatiche rimane la scintigrafia con Octreotide, un analogo della somatostatina, marcata con Indio 111. L’ecografia, la TAC e/o la risonanza magnetica addominali e la radiografia del torace permettono di identificare eventuali linfoadenomegalie e metastasi.
, metabolita della serotonina, deve essere dosato nelle urine delle 24 ore. Poiché la maggior parte dei C esprime recettori per la somatostatina, l’indagine di scelta per l’individuazione di lesioni residue extraddominali ed extraepatiche rimane la scintigrafia con Octreotide, un analogo della somatostatina, marcata con Indio 111. L’ecografia, la TAC e/o la risonanza magnetica addominali e la radiografia del torace permettono di identificare eventuali linfoadenomegalie e metastasi.")
50
Trattamento Esistono ancora controversie riguardo al trattamento più adeguato del CA, per il quale le dimensioni del tumore all’esame istologico sembrano rappresentare il fattore prognostico più importante. L’invasività locale (al mesenteriolo appendicolare ed al grasso periappendicolare) non viene ora considerato come un fattore prognostico sfavorevole in età pediatrica. In assenza di localizzazioni a distanza, l’osservazione dopo appendicectomia sembra essere la scelta adeguata per CA < 2 cm di diametro, con o senza infiltrazione della sierosa o del mesenteriolo. Tradizionalmente, l’emicolectomia destra è sempre stata considerata il trattamento di scelta per tumori di dimensioni > 2 cm per i quali sembra esistere un rischio di metastasi. Tuttavia un tale atteggiamento aggressivo non è condiviso dalla letteratura più recente: l’osservazione clinica ed umorale sembra essere l’opzione migliore anche in questi casi. L’asportazione del cieco con biopsia dei linfonodi pericolici deve essere eseguita nei pazienti con margini di resezione appendicolari non liberi da malattia..
 non viene ora considerato come un fattore prognostico sfavorevole in età pediatrica. In assenza di localizzazioni a distanza, l’osservazione dopo appendicectomia sembra essere la scelta adeguata per CA < 2 cm di diametro, con o senza infiltrazione della sierosa o del mesenteriolo. Tradizionalmente, l’emicolectomia destra è sempre stata considerata il trattamento di scelta per tumori di dimensioni > 2 cm per i quali sembra esistere un rischio di metastasi. Tuttavia un tale atteggiamento aggressivo non è condiviso dalla letteratura più recente: l’osservazione clinica ed umorale sembra essere l’opzione migliore anche in questi casi. L’asportazione del cieco con biopsia dei linfonodi pericolici deve essere eseguita nei pazienti con margini di resezione appendicolari non liberi da malattia..")
51
TUMORI DEL PANCREAS ESOCRINO
Il pancreatoblastoma e il tumore papillare solido-cistico del pancreas sono le due varietà di neoplasia che, anche se raramente, si possono osservare in età pediatrica. Il carcinoma pancreatico è stato riportato solo eccezionalmente nell’adolescente e per questa neoplasia l’approccio diagnostico-terapeutico non si differenzia da quello dell’adulto. Pancreatoblastoma Aspetti epidemiologici Il primo caso riconosciuto come PB per la presenza di caratteristici elementi squamosi risale al 1957 ed il primo studio istopatologico è stato presentato da Frable nel Solo nel 77 tuttavia, Horie pubblica una dettagliata descrizione di due casi ed utilizza per primo la definizione di pancreatoblastoma. Nell'ambito dei tumori primitivi del pancreas, il PB sembrerebbe presentarsi con una frequenza variabile compresa tra 0.16 e 0.5%. Nel 75% dei casi sono colpiti bambini di età inferiore agli 8 anni. In 7 casi è stato riscontrato alla nascita (associato in 3 pazienti a sindrome di Beckwith-Wiedemann) e recentemente è stato descritto un caso di PB cistico prenatale, associato anch'esso a sindrome di Beckwith-Wiedemann.
 e recentemente è stato descritto un caso di PB cistico prenatale, associato anch esso a sindrome di Beckwith-Wiedemann.")
52
Aspetti clinici e diagnostici
Il PB può manifestarsi come massa asintomatica o causare anoressia, perdita di peso, dolore. Talvolta possono essere presenti anche sintomi da ostruzione biliare. Il 35% dei pazienti presenta metastasi alla diagnosi che più frequentemente coinvolgono il fegato ed i linfonodi all’ilo epatico e splenico. Polmoni ed ossa sono più raramente coinvolti. Per estensione diretta, possono essere interessati l’omento, il peritoneo, la milza, il rene ed il surrene di sinistra. La produzione di alfa-fetoproteina (AFP) è segnalata frequentemente in letteratura, sia come aumento sierico (in più del 50% dei pazienti), che negli studi immunoistochimici sulla neoplasia. Se elevata alla diagnosi, può rappresentare un marker importante nel follow-up. Dal punto di vista radiologico, l’ecografia descrive una massa disomogenea, TAC ed RMN ne definiscono la sede, i limiti e le caratteristiche. Anche se gli aspetti radiologici possono essere d’aiuto, per giungere ad una diagnosi certa è comunque necessaria una biopsia che può essere ottenuta anche con ago sottile. Al patologo che esamina il materiale prelevato l’aspetto ricorda l’embriogenesi del pancreas, presumibilmente perché il tumore origina da cellule primordiali pancreatiche totipotenti.
 è segnalata frequentemente in letteratura, sia come aumento sierico (in più del 50% dei pazienti), che negli studi immunoistochimici sulla neoplasia. Se elevata alla diagnosi, può rappresentare un marker importante nel follow-up. Dal punto di vista radiologico, l’ecografia descrive una massa disomogenea, TAC ed RMN ne definiscono la sede, i limiti e le caratteristiche. Anche se gli aspetti radiologici possono essere d’aiuto, per giungere ad una diagnosi certa è comunque necessaria una biopsia che può essere ottenuta anche con ago sottile. Al patologo che esamina il materiale prelevato l’aspetto ricorda l’embriogenesi del pancreas, presumibilmente perché il tumore origina da cellule primordiali pancreatiche totipotenti.")
53
Trattamento e prognosi
Il comportamento del PB è quello di una neoplasia maligna ad alta aggressività. L’asportazione completa del tumore è necessaria per sperare in una prognosi favorevole. Questa tuttavia non è quasi mai fattibile alla diagnosi e può non essere possibile nemmeno dopo chemioterapia per le dimensioni e il coinvolgimento di organi vicini e per la necessità di eseguire una pancreatoduodenectomia. In tutti i casi, e comunque anche in quelli in cui sia stato possibile eseguire un’exeresi radicale, sembra essere opportuno un trattamento chemioterapico. Non è stato ancora identificato un regime terapeutico ottimale, tuttavia, una chemioterapia che includa cisplatino e antracicline, simile a quella utilizzata per l’epatoblastoma, sembra essere il trattamento più appropriato. Per quanto riguarda il trattamento radioterapico, sono stati descritti pochi casi, la maggior parte dei quali aveva ricevuto anche chemioterapia. In generale, nei casi in cui la radioterapia è stata adottata, si è assistito ad una riduzione del volume tumorale. La sopravvivenza a 5 anni rimane tuttavia insoddisfacente (più del 40% dei pazienti decede a causa della malattia) e la prognosi resta legata alla possibilità di una asportazione radicale ed all'assenza di metastasi.
 e la prognosi resta legata alla possibilità di una asportazione radicale ed all assenza di metastasi.")
67
aspirazione di corpi estranei è la causa della morte di più di 300 bambini ogni anno negli Stati Uniti. Anche se questo può verificarsi in qualsiasi fascia d'età, la maggioranza dei bambini sono di età compresa tra 1-3 anni. In un ampio rapporto recente, le arachidi sono state di gran lunga il comune aspirazione di corpo estraneo più. Questa è stata seguita da materiale organico, altre noci, popcorn, semi, oggetti di plastica e perni. Tosse, senso di soffocamento e / o dispnea sono presenti nella storia e risultati fisici di oltre il 90% dei bambini. Una radiografia del torace può essere utile, specie se l'ispirazione / film di scadenza è fatto. air trapping è la scoperta più comune radiografica. Solo una piccola percentuale di oggetti per via inalatoria sarà opaco sulle pellicole. Fluoroscopia è meno comunemente eseguita. La maggior parte dei corpi estranei finire in un bronco principale con una leggera preponderanza del lato destro.

72
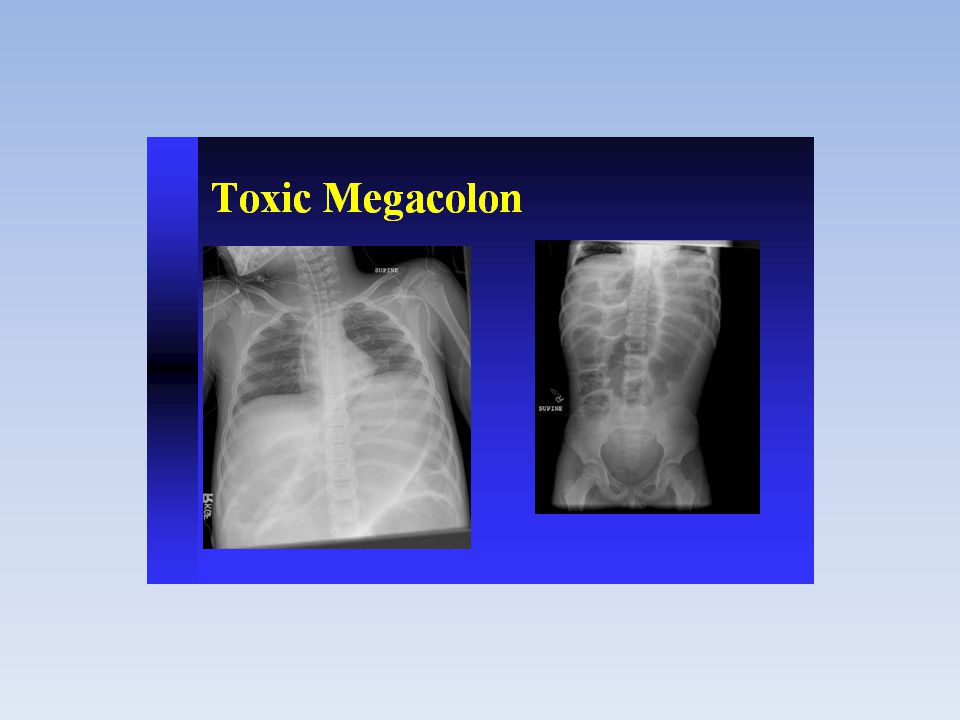
Hirschsprung’s Disease

73
Barium Enema

Presentazioni simili




















>")
















