Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Telethon Institute of Genetics and Medicine, Napoli
Cromosomopatie Vincenzo Nigro Laboratorio di genetica - Dipartimento di Patologia Generale, Seconda Università degli Studi di Napoli Telethon Institute of Genetics and Medicine, Napoli

2
Consulenza genetica La consulenza genetica è comunicazione informata ed appropriata Per essere informata deve partire dall’individuazione di un difetto genetico in un paziente e dal calcolo del rischio per gli altri componenti della famiglia Per poter essere appropriata deve saper stabilire un rapporto di fiducia e di confidenza senza essere direttiva, cioè non deve indirizzare la famiglia verso un unico obiettivo, ma lasciare libertà di valutazione e di scelta La consulenza genetica può riguardare: 1. la diagnosi di una malattia genetica clinicamente manifesta 2. il rischio riproduttivo di una coppia in epoca preconcezionale 3. la diagnosi prenatale 4. la predizione di una malattia genetica futura 5. la suscettibilità genetica

3
Consulenza genetica distinguiamo due grandi categorie di patologie genetiche: monoalleliche, dovute alla mutazione di una sola copia del DNA bialleliche, dovute a mutazioni di entrambe le copie del DNA patologie a penetranza completa (in genere disordini mendeliani) a penetranza incompleta, o addirittura “circoscritta”. La consulenza genetica cerca di stabilire quali membri della famiglia sono interessati ed eventualmente quali possono essere portatori, e quindi calcolare la probabilità di ogni altra persona nella famiglia (anche non ancora nata) di essere un portatore o di ereditare la malattia
 a penetranza incompleta, o addirittura circoscritta . La consulenza genetica cerca di stabilire quali membri della famiglia sono interessati ed eventualmente quali possono essere portatori, e quindi calcolare la probabilità di ogni altra persona nella famiglia (anche non ancora nata) di essere un portatore o di ereditare la malattia.")
4
rischio riproduttivo generale
per una coppia per cui l’anamnesi personale e familiare abbiano escluso un incremento del rischio rispetto alla popolazione è 3-5% in caso di difetti congeniti rilevabili alla nascita (anomalie cromosomiche 0.65%) 8-10% rilevabili entro i 10 anni di età
 8-10% rilevabili entro i 10 anni di età.")
5
I cromatidi restano associati mediante il centromero.
Durante la mitosi: ciascun cromosoma si duplica producendo due copie identiche: i cromatidi fratelli. I cromatidi restano associati mediante il centromero. le copie si separano. ciascuna copia migra in una cellula cromatidi fratelli centromero

6
LE FASI DELLA MITOSI Interfase Profase Metafase Anafase Telofase

7
meiosi La meiosi è il processo che porta alla formazione dei gameti
I gameti sono cellule aploidi: hanno la metà dei cromosomi delle cellule diploidi 23 è il numero di cromosomi dei gameti 46 è il numero di cromosomi di ogni altra cellula umana La meiosi consiste in due divisioni cellulari meiosi I (riduzionale) e meiosi II (equazionale) che producono quattro cellule aploidi
 e meiosi II (equazionale) che producono quattro cellule aploidi.")
8
meiosi Meiosi I Meiosi II Replicazione del DNA Divisione riduzionale
46 cromosomi 92 Cromatidi / 92 dsDNA Divisione riduzionale separazione delle coppie di cromosomi 23 cromosomi 46 cromatidi / 46 dsDNA Meiosi II Separazione dei cromatidi 23 cromatidi / 23 dsDNA

9
Meiosi I 46 cromosomi 92 Cromatidi / 92 dsDNA
Profase I: ciascun cromosoma si duplica e le due parti restano strettamente associate. Questi sono chiamati cromatidi fratelli. Il crossing-over avviene in questa fase Metafase I: I cromosomi omologhi si allineano al piano equatoriale Anafase I: Le coppie omologhe si separano e i cromatidi fratelli restano uniti Telofase I: le due cellule figlie contengono solo un cromosoma di ciascuna coppia

10
meiosi I, profase I Leptotene Zygotene Pachitene Diplotene Diacinesi
i cromosomi si rendono visibili Zygotene le coppie di cromosomi omologhi formano le tetradi Pachitene crossing over Diplotene i cromosomi iniziano a separarsi ma sono tenuti insieme dai chiasmi Diacinesi ulteriore accorciamento dei cromosomi omologhi

11
Meiosi II Profase II: il DNA delle due cellule figlie non si replica.
23 cromosomi 23 cromatidi / 23 dsDNA Profase II: il DNA delle due cellule figlie non si replica. Metafase II: i cromosomi si allineano al piano equatoriale Anafase II: I centromeri si dividono e i cromatidi fratelli migrano separatamente a ciascun polo Telofase II: la seconda divisione cellulare è completa. Quattro cellule figlie aploidi (23) sono ottenute
 sono ottenute.")
12
Gametogenesi Spermatogonium Oogonium Primary spermatocyte Primary
oocyte Secondary spermatocytes Secondary oocyte Gametogenesi Polar Body I 4 spermatids Polar Body II Fertilized Ovum 4 spermatozoa

13
maschio spermatogonio Per tutta la vita Mitosi alla pubertà
Oogonium spermatogonio Per tutta la vita Mitosi alla pubertà Primary oocyte spermatocita primario Meiosi in 64gg Secondary oocyte spermatocita Secondario maschio Polar Body I 4 spermatidi Polar Body II Fertilized Ovum 4 spermatozoi

14
femmina Periodo fetale alla nascita o prima dopo la nascita
dopo la pubertà Alla fertilizzazione Meiosi in progress Si arresta al diplotene della meiosi I Meiosi I completa Si arresta alla metafase II Meiosi II Oogonio Mitosis oocita primario femmina oocita secondario e corpo polare I Fertilized Ovum & Polar body II

15
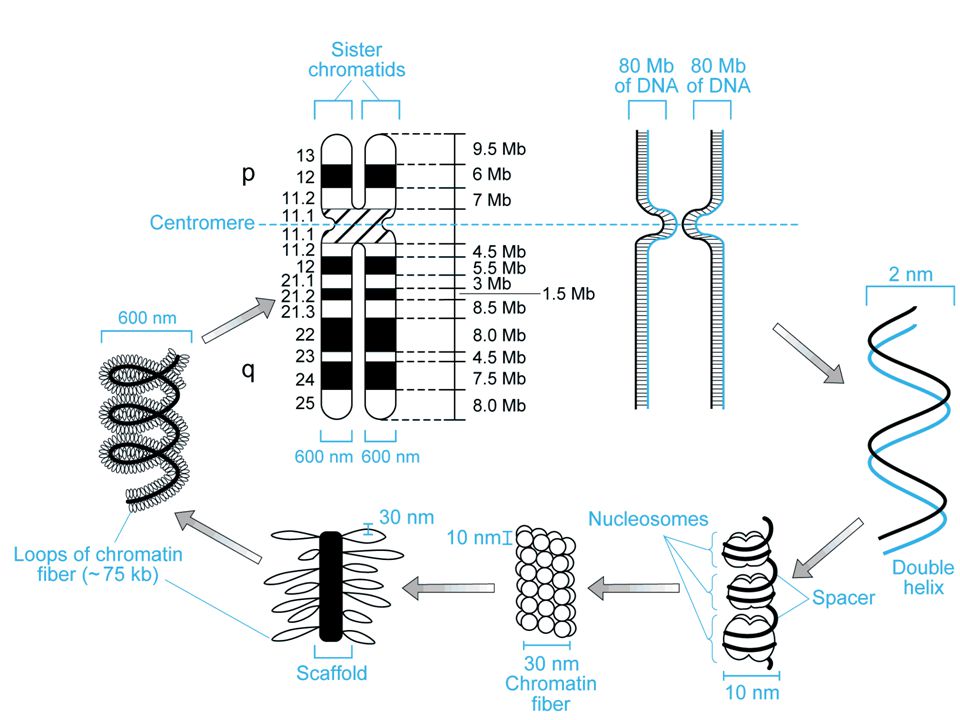
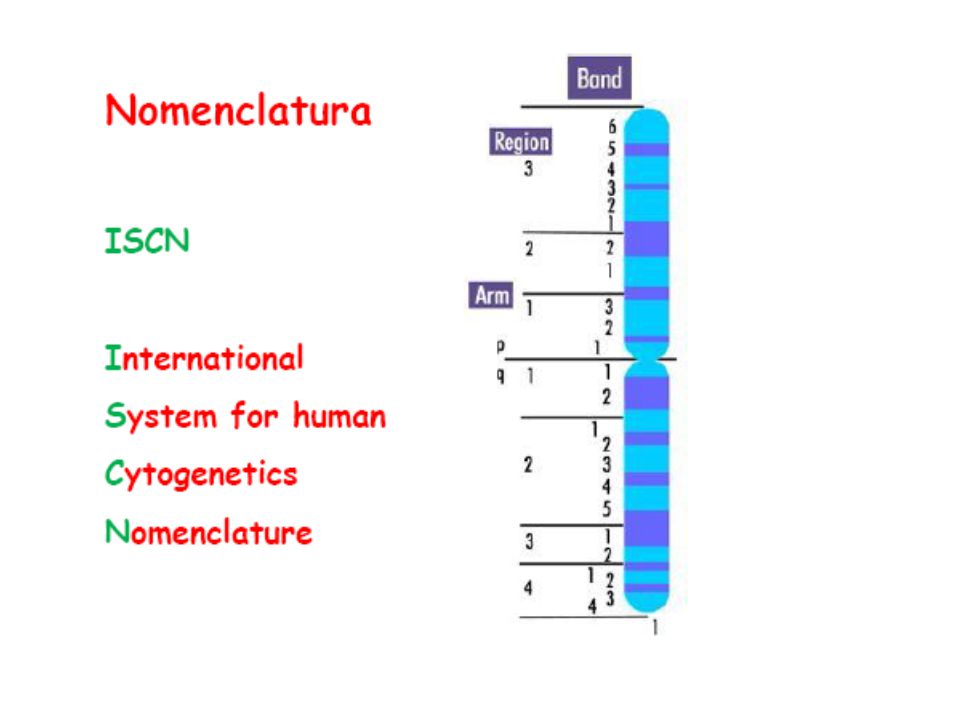
Cromosomi (corpi colorati)
durante il ciclo cellulare i cromosomi replicano e si formano due cromatidi fratelli tenuti insieme dal centromero braccio corto = p (petit) braccio lungo = q (lettera successiva)
 braccio lungo = q (lettera successiva)")
17
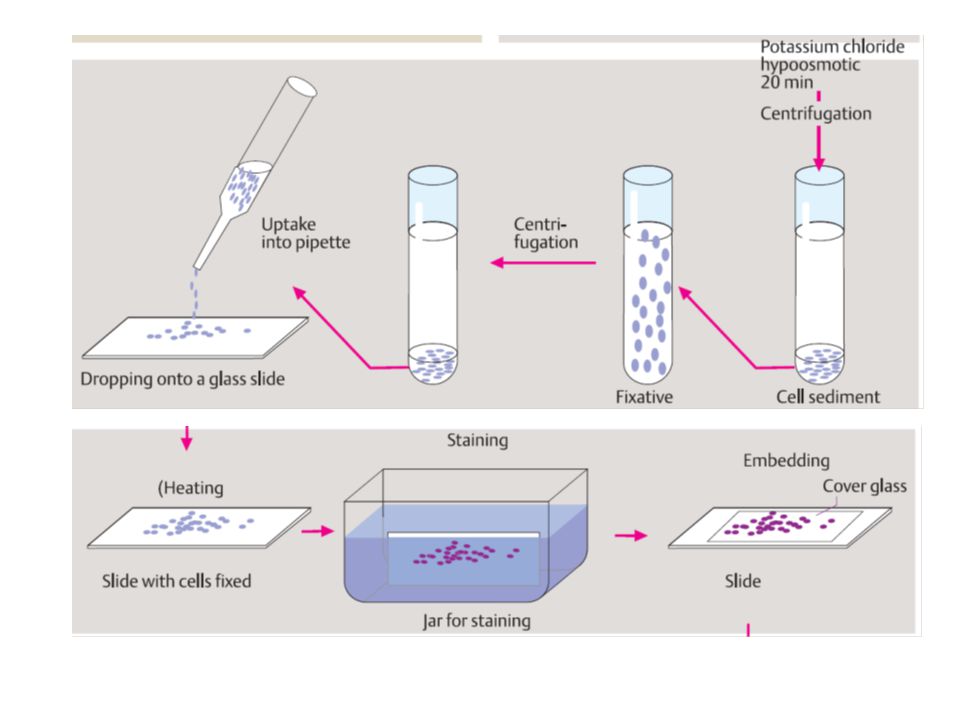
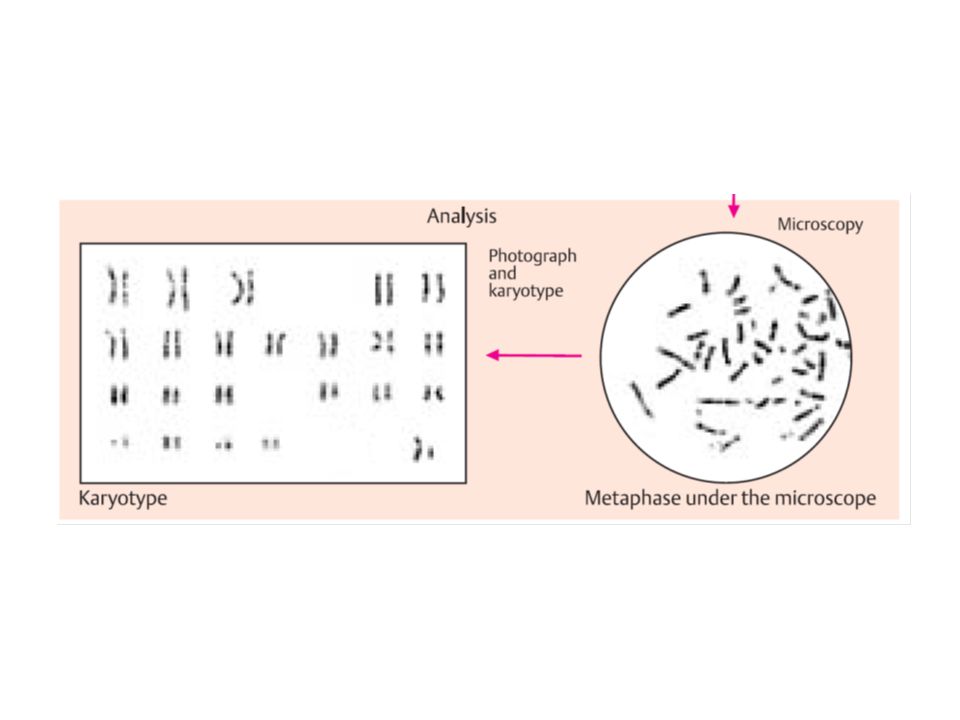
citogenetica di routine
da linfociti sono rappresentativi di ciascun altra cellula del corpo

19
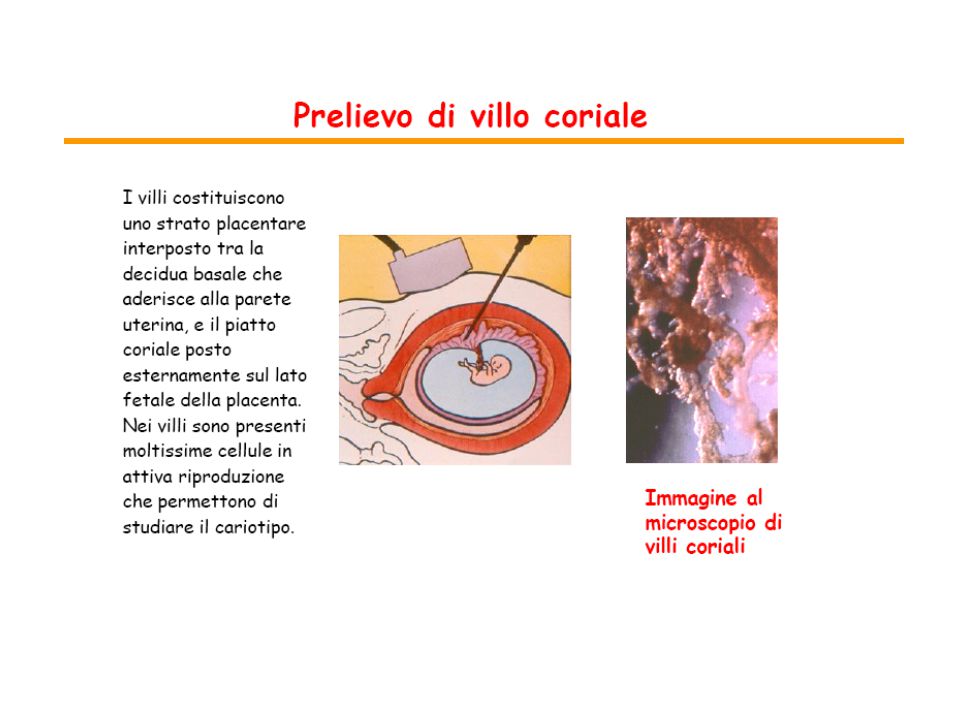
citogenetica prenatale
da amniociti da villi coriali dovrebbero essere rappresentativi delle cellule del feto difficili da ottenere

21
Cromatina (DNA+proteine)
Eucromatina - meno condensata contiene il DNA codificante Eterocromatina - più condensata non contiene DNA codificante, ma solo DNA non codificante Telomeri - cappucci all’estremità dei cromosomi che comprendono ripetizioni multiple della sequenza TTAGGG Centromeri - regioni specializzate di DNA che forniscono il sito di ancoraggio del fuso mitotico

22
Eucromatina ed eterocromatina

23
Tecnica Procedura Banding pattern bandeggio G Proteolisi limitata seguita dalla colorazione Giemsa Le bande scure sono ricche in AT Le bande chiare sono ricche in GC bandeggio R denaturazione al calore seguita dalla colrazione con Giemsa Le bande scure sono ricche in GC Le bande chiare sono ricche in AT bandeggio Q digestione enzimatica e colorazione con un colorante fluorescente, cioè la Quinacrina bandeggio C denaturazione con idrossido di bario e poi colorazione con Giemsa Le bande scure sono ricche in eterocromatina costitutiva

24
Colorazione dei cromosomi con coloranti specifici
per regioni ricche in AT o in GC

26
metacentrici, se il centromero è centrale 1, 2, 3, 16, 17, 18, 19
submetacentrici, se il centromero non è centrale e non è vicino ad un’estremità 4, 5, 6, 7, 8, 9, 10, 11, 12, 20, X, Y acrocentrici, se il centromero è vicino ad un’estremità 13, 14, 15, 21, 22

28
CCDS IDs per chromosome
Count 1 2,513 2 1,548 3 1,299 4 898 5 1,028 6 1,236 7 1,094 8 807 9 921 10 971 11 1,509 12 1,240 13 385 14 749 15 711 16 967 17 1,370 18 350 19 1,616 20 672 21 282 22 530 X Y 53 XY 23

29
Eteromorfismi citogenetici
Variazione pericentromerica del crom. 9 9qh+ Inversione 9 inv Variazione + inversione

30
Ereditarietà della variazione pericentromerica del cromosoma 1

31
Le alterazioni cromosomiche sono più frequenti al crescere dell’età materna, mentre le mutazioni puntiformi sono legate al numero di divisioni cellulari che avvengono circa ogni 15 gg nella linea germinale maschile

32
tritest interpretazione dei risultati
anomalia fetale AFP Alfa-feto proteina Beta hCG uE estriolo non coniugato NTD =difetti del tubo neurale* Normale Trisomia 21 18 * NTD: anencefalia, spina bifida and encefalocele

33
Il duotest (double screen) include la valutazione del PAPP-A (Pregnancy Associated Plasma Protein A) e la frazione libera della gonadotropina corionica (free-betaHCG). Viene effettuato tra la 10ma e la 13ma settimana di gravidanza dal siero della gestante Translucenza nucale free-bHCG PAPP-A Trisomia 21 ++ - Trisomia 13,18 +++ - - S. di Turner ++++ +/- Triploidia materna - - - Triploidia paterna

34
Patologia fetale Sensibilità NTD - AFP solo 75-80% spina bifida 95% anencefalia Trisomia 21 - Tritest 70% Down sindrome Trisomia 18 - Tritest 80% sindrome di Edward

35
Anomalie ecografiche maggiori

36
ecografia “segni minori”

37
Frequenza di anomalie cromosomiche negli aborti spontanei (39. 8%-40
trisomie autosomiche 49-52% Turner (45, X) 15-19% triploidia (69) 15-16% tetraploidia (92) 5-6% altre anomalie 6-14%
 15-19% triploidia (69) 15-16% tetraploidia (92) 5-6% altre anomalie 6-14%")
38
trisomia 21 Down Il 70% delle gravidanze non giunge a termine

39
1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 1 1 2 2 Meiosis 1 Meiosis 1 error Meiosis 1 Meiosis 1 1 1 1 1 2 2 1 1 1 1 + 1 2 1 2 1 2 1 2 1 2 Meiosis 2 Meiosis 2 error Meiosis 2 Meiosis 2 1 1 1 2 1 1 1 or + or 3 other combinations + or other combinations Mitotic error 1 2 1 1 2 2 1 1 1 2 2 SEA3069

40
origine dell’extra cromosoma 21
MM2 19.8% PM1 2.6% MM1 68% PM2 4.1% MIT 5.5% Data from the Antonarakis and Hassold laboratories sea3109

41
anomalie cromosomiche riscontrate
13 12 11.2 p 11.1 27 11.1 mosaicism 11.2 2 altre t 3 t21;22 17 21 t21;21 5 Anomalia t15;21 q 6 t13;21 15 22.1 t14;21 925 free T21 D21S17 22.2 DSCR 1 ETS2 10 100 1000 22.3 Numero MX1 HC21

42
Ipotesi sulla variabilità del fenotipo di 6 individui diversi in caso di trisomia 21 in presenza di varianti alleliche fenotipo Livello di espressione

43
trisomia 21 Down 40.000 casi in Italia Neurologici :
Ritardo mentale 100% Alzheimer dopo i 35anni 100% Ipotonia muscolare 100% Bassa statura 70% Testa : Brachicefalia 75% Epicanto 60% brushfield spots iride 55% lingua protrudente 45% orecchie displastiche 50%

44
trisomia 21 Down Arti corti, mani larghe 65% Mignolo corto 60%
Solco palmare trasverso 60% Cuore Difetti cardiaci congeniti 40% Canale atrioventricolare 16% Anomali gastrointestinali Atresia/stenosi duodenale 250x ano imperforato 50x Hirschsprung 300x Sangue: Leucemia acuta megacariocitica 300x Leucemia (ALL e AML) x
 10-20x.")
45
trisomia 18 Edwards (1/6.500 nati)
90% dei casi nondisgiunzione materna M/F = 1/4 Giunge a termine solo il 2.5% dei concepimenti Di questi il 33% muore nel primo mese, il 50% entro 2 mesi Oltre 100 anomalie Peso sotto la norma, difficoltà suzione Ipotonia Idrocefalo, epilessia Malformazioni cardiache sinclinodattilia, unghie poco sviluppate piedi a calcagno prominente Gambe incrociate

46
trisomia 13 Patau (1/12.000-20.000 nati)
(1/ nati) 90% dei casi nondisgiunzione materna Giunge a termine solo il 2.5% dei concepimenti Di questi il 33% muore nel primo mese, il 50% entro 2 mesi Peso sotto la norma, difficoltà suzione Oloprosencefalia, microcefalia Cecità e sordità Occhi che possono fondersi Labiopalatoschisi 80% epilessia Malformazioni cardiache sinclinodattilia piedi a calcagno prominente
 90% dei casi nondisgiunzione materna. Giunge a termine solo il 2.5% dei concepimenti. Di questi il 33% muore nel primo mese, il 50% entro 2 mesi. Peso sotto la norma, difficoltà suzione. Oloprosencefalia, microcefalia. Cecità e sordità. Occhi che possono fondersi. Labiopalatoschisi 80% epilessia. Malformazioni cardiache. sinclinodattilia. piedi a calcagno prominente.")
47
XX o XY Il sesso maschile è determinato dalla presenza del cromosoma Y
Si sono evoluti meccanismi per compensare la differenza di dosaggio genico del cromosoma X, presente in 2 copie nelle femmine e in 1 copia nei maschi

48
2 cromosomi X nelle donne,
1 solo negli uomini? il cromosoma X raddoppia l’espressione di tutti i geni contenuti, cioè si produce 2 volte più RNA nelle femmine uno dei due cromosomi X è inattivato casualmente in ciascuna cellula allo stadio di blastocisti

49
Il Klinefelter? Nel Klinefelter (XXY) uno dei due cromosomi X è inattivato casualmente in ciascuna cellula allo stadio di blastocisti Quindi il dosaggio sarebbe mantenuto

50
Sindrome di Klinefelter (47,XXY) 1:900-1:600 maschi
Il 50% delle gravidanze giunge a termine Fenotipo maschile Caratteristiche principali: Statura alta Ipogonadismo, bassi livelli di testosterone, mancata produzione di spermatozoi (azoospermia) e quindi sterilità Ginecomastia Sia l’intelligenza sia l’attesa di vita sono quasi normali
 e quindi sterilità. Ginecomastia. Sia l’intelligenza sia l’attesa di vita sono quasi normali.")
51
Altre forme citogenetiche
Ma ci sono anche Klinefeler 48,XXYY and 48,XXXY in 1 caso su 17,000 e 1 su 50,000 mnati maschi 49,XXXXY in 1 caso su 85, ,000 Ci sono maschi 46,XX in cui avviene una traslocazione di parte di cromosoma Y sul cromosoma X che include la sex determining region (SRY) mosaici
 mosaici.")
52
PAR1 ha 24 geni, PAR2 ha solo 4 geni
Le regioni PAR presenti sui cromosomi sessuali contengono geni che non sono inattivati, perché il doppio dosaggio è assicurato comunque PAR1 ha 24 geni, PAR2 ha solo 4 geni

53
Il gene SHOX Short stature HOmeoboX-containing
Mutazioni o delezioni del gene SHOX nella regione PAR1 causa ritardo di crescita e bassa statura. La bassa statura di donne con sindrome di Turner Syndrome (X0) è il risultato di una sola copia di SHOX (ma anche il quarto metacarpo corto) La maggiore statura nel Klinefelter (XXY) e nella tripla X (XXX) potrebbe essere il risultato di 3 copie di SHOX
 è il risultato di una sola copia di SHOX (ma anche il quarto metacarpo corto) La maggiore statura nel Klinefelter (XXY) e nella tripla X (XXX) potrebbe essere il risultato di 3 copie di SHOX.")
54
3 copie nel Klinefelter, ma anche nella tripla X
variabilità dei geni del cromosoma X delle regioni PAR, quindi non inattivati 3 copie nel Klinefelter, ma anche nella tripla X Mario Rossi Luca Bianchi Pio Verdi Giulio Rosa Lucio Viola Gianni Neri Livello di espressione

55
A complicare le cose… circa il 15 % dei geni umani presenti sull’X sfugge all’inattivazione, mentre nel topo questa è un’evenienza rara (solo 6 geni in tutto) alcuni sono espressi al % altri al 10% questo fenomeno è quindi incompleto e le donne hanno una elevata eterogeneità nell’espressione di geni dell’X
 alcuni sono espressi al % altri al 10% questo fenomeno è quindi incompleto e le donne hanno una elevata eterogeneità nell’espressione di geni dell’X.")
56
Manifestazione clinica: NO Manifestazione clinica: SI
ipotesi sulla variabilità di ogni singola manifestazione clinica di Klinefelter in presenza di varianti in geni del cromosoma X non inattivati Manifestazione clinica: NO Manifestazione clinica: SI Mario Rossi Luca Bianchi Pio Verdi Giulio Rosa Lucio Viola Gianni Neri Livello di espressione

57
Quanti Klinefelter? Prevalenza di XXYs è cresciuta da 1.09 a 1.72 per 1000 maschi nati (P=0.023) Questo incremento non è dovuto all’aumento dell’età materna Sono nati maschi in Italia e maschi in Campania nel 2007 max nuovi Klinefelter ogni anno in Italia (32-52 in Campania) XXY è la sola trisomia nota in cui circa il 50% dei casi è causato da una non disgiunzione alla prima divisione meiotica paterna
 XXY è la sola trisomia nota in cui circa il 50% dei casi è causato da una non disgiunzione alla prima divisione meiotica paterna.")
58
Trisomia X (47,XXX) 1:1.200 Il 70% delle gravidanze giunge a termine
Errore nella disgiunzione materna e correla con l’età materna Caratteristiche principali: Statura alta Fertilità normale, irregolarità ciclo Sia l’intelligenza sia l’attesa di vita sono normali

59
Maschio (47,XYY) 1:1.000 maschi Fenotipo maschile
Caratteristiche principali: Statura alta Fertilità normale Non vi è correlazione con l’età paterna Sia l’intelligenza sia l’attesa di vita sono perfettamente normali

60
Monosomia X (45,X0) Turner Prende il nome dall’endocrinologo Henry Turner che la descrisse nel 1938 La sindrome di Turner (TS) definisce un complesso fenotipo umano femminile, dovuto a completa o parziale assenza del secondo cromosoma sessuale Dipende da un errore nella spermatogenesi nell’80% dei casi e non correla con l’età dei genitori Un precedente figlio con TS non aumenta il rischio riproduttivo previsto per una coppia di pari età
 definisce un complesso fenotipo umano femminile, dovuto a completa o parziale assenza del secondo cromosoma sessuale. Dipende da un errore nella spermatogenesi nell’80% dei casi e non correla con l’età dei genitori. Un precedente figlio con TS non aumenta il rischio riproduttivo previsto per una coppia di pari età.")
61
Monosomia X (45,X0) Turner È l’unica monosomia compatibile con la vita, ma il 98% di tutti i feti monosomici TS va incontro ad aborto spontaneo L’incidenza negli aborti è circa il 7-10%, mentre alla nascita è 1/2500 femmine. Non è chiaro perché il cariotipo 45, X0 sia letale in utero ed invece compatibile con la sopravvivenza postnatale La vera monosomia del cromosoma X è responsabile del 45% dei casi TS; gli altri hanno mosaicismo (45, X0/46, XX) e/o un anormale cromosoma X o Y Un basso livello di mosaicismo somatico Turner, inferiore al 2%, è di normale riscontro nella popolazione
 e/o un anormale cromosoma X o Y. Un basso livello di mosaicismo somatico Turner, inferiore al 2%, è di normale riscontro nella popolazione.")
62
Monosomia X (45,X0) Turner “la menopausa precede il menarca”
Le ovaie sono allungate e formate da tessuto stromale privo di follicoli:gli oociti sono spesso andati in apoptosi prima dei 2 anni di vita L’insufficienza ovarica prepuberale porta ad amenorrea primaria, sterilità e carenza di estrogeni In meno del 10% dei casi, la pubertà può verificarsi e sono possibili gravidanze con un aumentato rischio di perdita fetale Anche in rapporto all’eterogeneità del genotipo, il fenotipo si manifesta in modo molto variabile

63
Monosomia X (45,X0) Turner 1:2.500
patologie dell’orecchio medio (otite media ricorrente) Linfedema con rigonfiamento delle mani e dei piedi pterigio del collo (presenza di pliche cutanee con aspetto di sfinge) il quarto metacarpo (anulare) più corto una mandibola più piccola (micrognazia) torace largo con aumento degli spazi intercostali l’attaccatura bassa delle orecchie e dei capelli Si possono anche riscontrare cardiopatia sinistra (valvola aortica dicuspide, coartazione aortica), ipertensione e anomalie renali
 Linfedema con rigonfiamento delle mani e dei piedi. pterigio del collo (presenza di pliche cutanee con. aspetto di sfinge) il quarto metacarpo (anulare) più corto. una mandibola più piccola (micrognazia) torace largo con aumento degli. spazi intercostali. l’attaccatura bassa delle orecchie e dei capelli. Si possono anche riscontrare cardiopatia sinistra (valvola aortica dicuspide, coartazione aortica), ipertensione e anomalie renali.")
64
feto con anomalia cromosomica (mosaicismo)
trisomie a mosaico 8, 9, 13, 18, 21 crescita in coltura di cellule materne mosaicismo vero (livello III) pseudomosaicismo (livelli II e I)
 pseudomosaicismo (livelli II e I)")
65
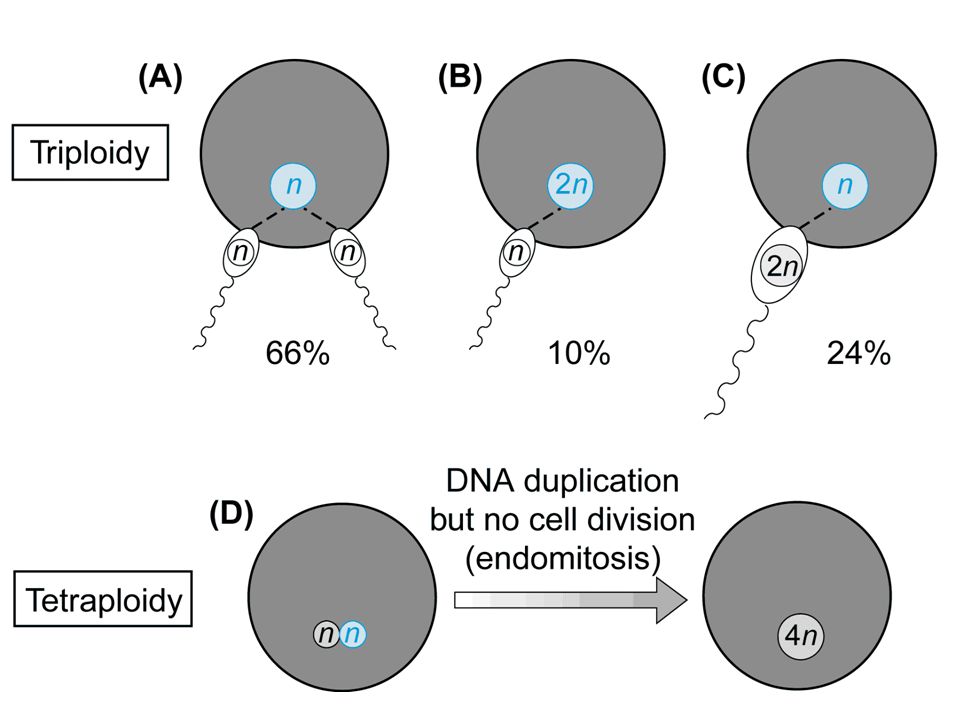
triploidia Frequenza alla nascita = 1/10.000
Frequenza negli aborti = 1/14 Cariotipo 69,XXY 57% Cariotipo 69,XXX 40% Cariotipo 69,XYY 3%

68
Nati vivi Tipo I, corredo sovrannumerario paterno
Feto microcefalico o normale Placenta ingrossata Tipo II, corredo sovrannumerario materno Ritardo di crescita Feto con macrocefalia relativa Placenta poco sviluppata Nati vivi Basso peso Asimmetria cranio-facciale e difetti di ossificazione del cranio Microftalmia, ipertelorismo, micrognazia Sindattilia cutanea, piedi torti Anomalie genitali, ipoplasia delle surrenali Cardiopatie

69
Mosaicismo, quando il cambiamento avviene dopo che si è formato lo zigote
47,XXY/46,XY

70
Un precedente figlio con anomalie cromosomiche
Aumenta il rischio in caso di: tutte le trisomie non mosaico riarrangiamenti strutturali marker cromosomi

71
Un precedente figlio con anomalie cromosomiche
NON aumenta il rischio in caso di: 47, XYY triploidia, tetraploidia sindrome di Turner

72
Valutazione del rischio riproduttivo nel periodo preconcezionale
momento ottimale (ma oltre la metà delle gestazioni insorge inaspettatamente) raccolta dei dati (visita, abitudini, terapie, accertamenti lab) SCOPO: identificazione dei portatori sani di malattie genetiche portatori che hanno un rischio riproduttivo a prescindere dal partner portatori in cui il rischio si manifesta solo nel caso di unione con un partner portatore
 raccolta dei dati (visita, abitudini, terapie, accertamenti lab) SCOPO: identificazione dei portatori sani di malattie genetiche. portatori che hanno un rischio riproduttivo a prescindere dal partner. portatori in cui il rischio si manifesta solo nel caso di unione con un partner portatore.")
73
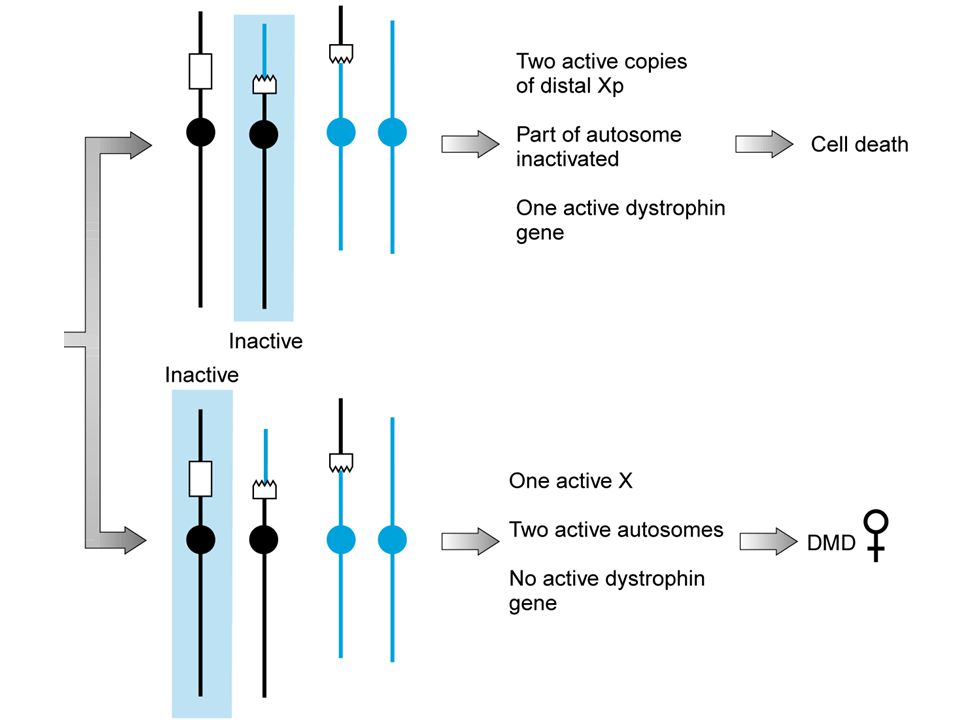
portatori che hanno un rischio riproduttivo a prescindere dal partner
donne con mutazioni legate all’X (esempio: Distrofia muscolare di Duchenne)
")
74
portatori che hanno un rischio riproduttivo a prescindere dal partner
reciproca portatori di una traslocazione cromosomica bilanciata scambio di materiale genetico tra cromosomi non omologhi non vi è modificazione della dose genica frequenza 1/520 nati fenotipicamente normale

75
traslocazioni X-autosoma traslocazioni robertsoniane
coppia con familiarità per anomalie cromosomiche è indicazione all’esecuzione di un cariotipo fetale e l’estensione dell’indagine ai parenti traslocazioni X-autosoma maschi sterili, femmine inattivano la X normale traslocazioni robertsoniane non 21 60% cariotipo bilanciato 21 15% rischio di Down, se è eterozigote la madre 1% se è eterozigote il padre inversioni pericentriche varianti dell’1, 9, 16 e Y, in altri casi il rischio è 5-10% paracentriche, rischi inferiore allo 0.5%

76
Traslocazioni bilanciate (meiosi e fertilizzazione)
Traslocazione bilanciata Segregazione alternata Normale Traslocazione Segregazione adiacente 1 Traslocazione Trisomia Segregazione adiacente 2 Trisomia

77
influenze sugli effetti della traslocazione
Cromosomi coinvolti e lunghezza del tratto traslocato (perché vi è una forte selezione pre e postzigotica): maggiori sono le dimensioni cromosomiche minore è la possibilità di una gravidanza a termine Sesso del genitore donna>uomo (gli spermatozoi hanno il 7.5% di difetti contro l’1% degli oociti, ma sono selezionati) Il rischio aumenta se il difetto è stato accertato a partire da un figlio precedente con cariotipo sbilanciato
: maggiori sono le dimensioni cromosomiche minore è la possibilità di una gravidanza a termine. Sesso del genitore donna>uomo (gli spermatozoi hanno il 7.5% di difetti contro l’1% degli oociti, ma sono selezionati) Il rischio aumenta se il difetto è stato accertato a partire da un figlio precedente con cariotipo sbilanciato.")
78
rischio alla nascita di figli con cariotipo sbilanciato
Se non vi sono stati casi in famiglia e la madre è eterozigote per una traslocazione reciproca il rischio è il 7% Se non vi sono stati casi in famiglia e il padre è eterozigote per una traslocazione reciproca il rischio è il 3% Se vi sono stati casi di traslocazioni sbilanciate in famiglia e la madre è eterozigote il rischio è il 14% Se vi sono stati casi di traslocazioni sbilanciate in famiglia e il padre è eterozigote il rischio è l’8%

79
donna eterozigote per una traslocazione bilanciata X-autosoma

81
Traslocazioni reciproca Robertsoniana

82
t(13;14) M=F 1% t(14;21) F 15% M 2% t(21;22) F 10% M 5%
Rischio alla nascita di figli con cariotipo sbilanciato da genitori con traslocazione robertsoniana t(13;14) M=F 1% t(14;21) F 15% M 2% t(21;22) F 10% M 5% t(21;21) M=F 100%
 M=F 1% t(14;21) F 15% M 2% t(21;22) F 10% M 5% t(21;21) M=F 100%")
83
portatori che hanno un rischio riproduttivo a prescindere dal partner
mutazioni dominanti ad esordio tardivo (corea di Huntington, atassie spinocerebellari) mutazioni dominanti a penetranza incompleta
 mutazioni dominanti a penetranza incompleta.")
84
portatori in cui il rischio si manifesta solo nel caso di unione con un altro portatore
mutazioni autosomiche recessive con familiarità (coppia già a rischio) senza familiarità (valutare la consanguineità)
 senza familiarità (valutare la consanguineità)")
85
ogni individuo è portatore sano di almeno 8 malattie genetiche recessive, di cui 3 letali
fratelli, genitori-figli fratellastri, zii-nipoti cugini diretti (0.5%) secondi cugini 1/4 omozigosi 1/8 omozigosi 1/16 omozigosi 1/64 omozigosi i difetti congeniti hanno un rischio empirico raddoppiato in caso di cugini primi non è utile l’esame cromosomico
 secondi cugini. 1/4 omozigosi. 1/8 omozigosi. 1/16 omozigosi. 1/64 omozigosi. i difetti congeniti hanno un rischio empirico raddoppiato in caso di cugini primi non è utile l’esame cromosomico.")
87
feto con anomalia cromosomica (mosaicismo)
trisomie a mosaico 8, 9, 13, 18, 21 crescita in coltura di cellule materne mosaicismo vero (livello III) pseudomosaicismo (livelli II e I)
 pseudomosaicismo (livelli II e I)")
88
Whitehead Institute, Center for Genome Research, Cambridge, MA

90
La grandezza totale del genoma umano aploide è di 3. 070. 000
La grandezza totale del genoma umano aploide è di basi di cui sono di eucromatina

91
UCSC Genome Browser Screenshot from University of California at Santa Cruz

92
disordine genomico submicroscopico
Il disordine genomico submicroscopico è una patologia causata da acquisizione perdita alterazione di uno o più geni contigui le cui variazioni di dosaggio possono produrre effetti fenotipici La base molecolare è rappresentata da riarrangiamenti genomici, quali duplicazioni, delezioni, inversioni, senza grosse alterazioni del cariotipo (<5Mb)
")
93
In caso di delezioni del cromosoma X nei maschi si osserva direttamente in fenotipo come sindrome da geni contigui In caso di delezioni autosomiche in eterozigosi, molto spesso il dosaggio dimezzato non è causa di malattia. Quando si osserva una sindrome da delezione, è risolutivo trovare la stessa sindrome causata da una mutazione puntiforme in uno solo dei geni. Se questa non si trova, la sindrome esiste solo come somma di più difetti.

94
ACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGAACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGA

95
10% of the human genome could vary in copy number
ACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGAACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACTATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGATAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTTATAGCTCGCGACACACACAGATATATAGCGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGACGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTGACCTGACACGTGCTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGACACACACAGATATATAGCGCTCCCTGAAACAGCTCCGACACAGCTCGCACACCGCTCGAGACCTTAGCTAGCTCCTCTCGAGACGTAGGGCTCTCGATATAGCTCGCGA Copy Number Variation 10% of the human genome could vary in copy number 1 2

96
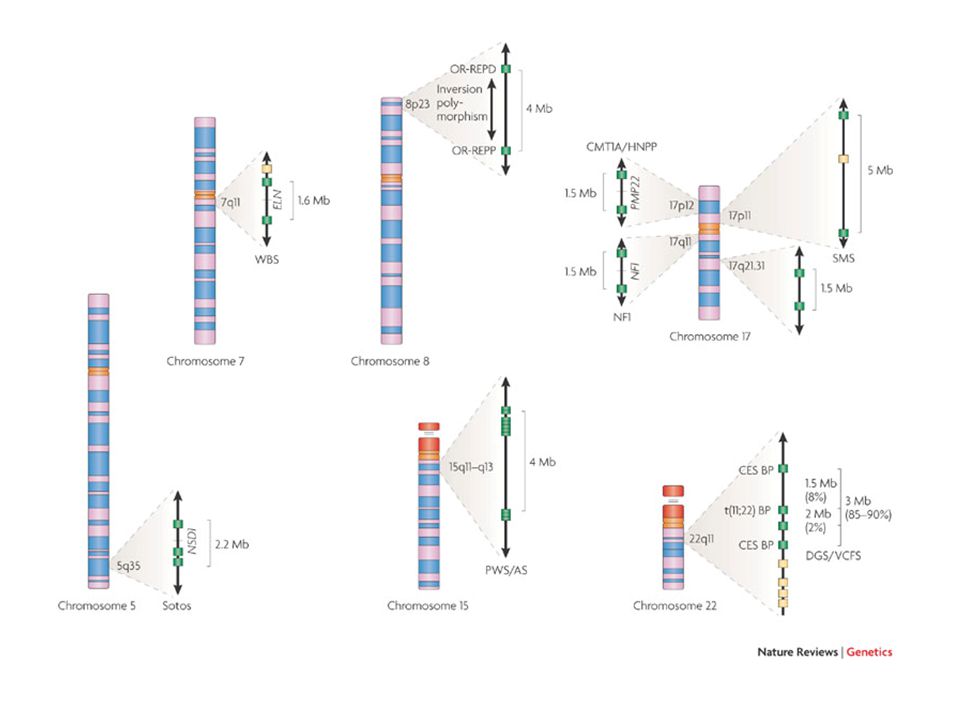
principali sindromi da delezione

97
FISH

99
Sonde FISH subtelomero centromero intero cromosoma locus

100
PAINTING

103
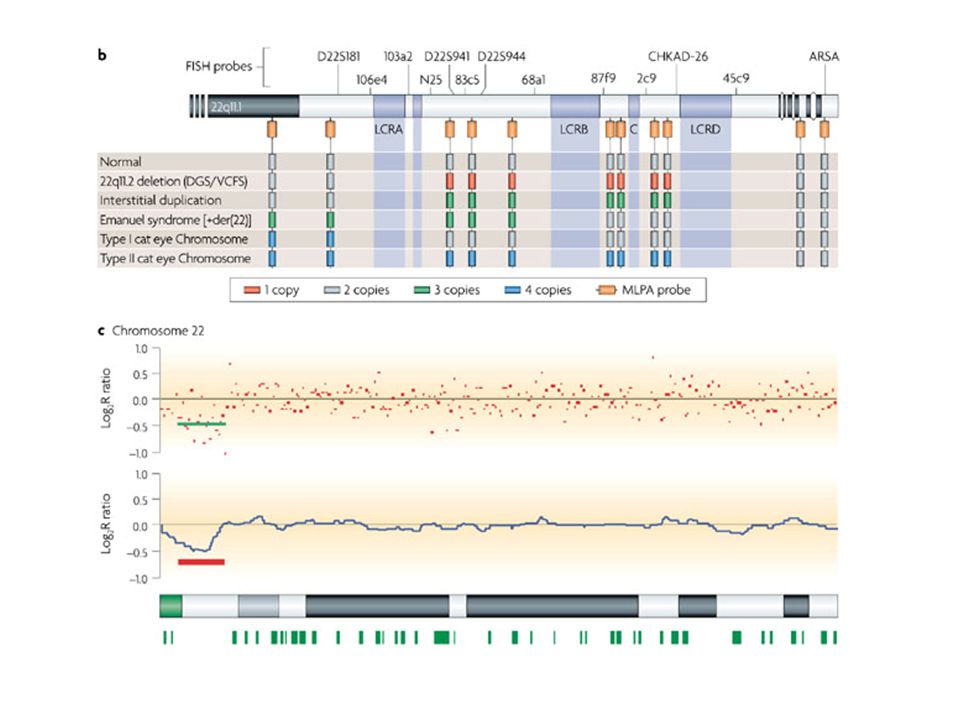
Sindrome di DiGeorge

104
DiGeorge/velocardiofacciale
La sindrome di DiGeorge del22q11.2 è la più frequente sindrome da microdelezione, con un incidenza di 1 su 4000—5000 nati La delezione comprende 3Mb ed almeno 30 geni

105
Migrating neural crest cells make a contribution to the embryonic structures affected in DiGeorge syndrome. The cartoon represents a human embryo at 4–6 weeks gestation. The migration of neural crest cells from the hindbrain to the branchial arch/pharyngeal pouch system and cardiac outflow tract is indicated by the arrows. Examples of malformations associated with perturbation of this process are listed and these overlap substantially with those seen in 22q11DS AAA, arch arteries; PDA, persistent ductus arteriosus; IAA, interrupted aortic arch.

106
DiGeorge È caratterizzata da Anomalie cardiache T-cell deficit
palatoschisi anomalie facciali Ipocalcemia Mutazioni puntiformi del gene TBX1 possono portare a questi 5 tratti fenotipici, ma non alle difficoltà nell’apprendimento che è invece frequente nella sindrome da delezione

107
Williams-Beuren prevalenza alla nascita 1/7500-1/20.000, ma può non essere diagnosticata

108
Williams una delezione tipica

109
Williams genetica gene dell’elastina LIM kinase 1 (LIMK1)
delezione “de novo” trasmissione autosomica dominante delezione di 1.6MB da 21 geni contigui in eterozigosi a 7q11.23 gene dell’elastina LIM kinase 1 (LIMK1) CLIP-115 che lega i microtubuli Fattori di trascrizione GTF2I e GTF2IRD1 effetto posizionale su altri geni circostanti la delezione
 CLIP-115 che lega i microtubuli. Fattori di trascrizione GTF2I e GTF2IRD1. effetto posizionale su altri geni circostanti la delezione.")
110
Williams FISH delezione 7q11.23
rilevabile mediante FISH ma non cariotipo

111
Williams comportamento
lieve o medio ritardo mentale (IQ tra 41 e 80) scarsa capacità di concentrazione ritardo nell’apprendimento del linguaggio e poi esagerata loquacità personalità amichevole e affettuosa danno facilmente confidenza anche a sconosciuti ansietà, spesso preoccupati per il benessere altrui ipersensibilità ai suoni memoria visiva e uditiva spesso fuori dal comune ricordano persone, luoghi e motivi musicali predisposizione ad imparare le lingue e la musica
 scarsa capacità di concentrazione. ritardo nell’apprendimento del linguaggio e poi esagerata loquacità. personalità amichevole e affettuosa. danno facilmente confidenza anche a sconosciuti. ansietà, spesso preoccupati per il benessere altrui. ipersensibilità ai suoni. memoria visiva e uditiva spesso fuori dal comune. ricordano persone, luoghi e motivi musicali. predisposizione ad imparare le lingue e la musica.")
112
Williams aspetto e segni
Faccia da elfo Occhi blu (77%) con pattern stellato dell’iride (74%) ma questo vale per i nordeuropei, strabismo (40%) Naso con la punta bulbosa bocca larga e guance piene microdontia e micrognazia Statura 10 cm in meno del normale ipercalcemia stenosi periferica delle arterie polmonari stenosi aortica sopravalvolare
 con pattern stellato dell’iride (74%) ma questo vale per i nordeuropei, strabismo (40%) Naso con la punta bulbosa. bocca larga e guance piene. microdontia e micrognazia. Statura 10 cm in meno del normale. ipercalcemia. stenosi periferica delle arterie polmonari. stenosi aortica sopravalvolare.")
113
Williams foto

114
Williams foto

115
Wolf-Hirschhorn genetica
delezione “de novo” di circa 4MB le delezioni sono più frequenti nella linea germinale maschile trasmissione autosomica dominante Regione critica di 165 kb di molti geni contigui in eterozigosi a 4p16.3

116
Wolf-Hirschhorn delezione a 4p16.3

117
Wolf-Hirschhorn Scarso accrescimento Ritardo mentale, ipotonia
Labbro leporino Conformazione ad elmo di guerriero greco

118
Sindrome 5p- (cri du chat) 1:50.000 nati
Pianto acuto e flebile Caratteristiche principali: Ritardo di crescita Microcefalia ed ipertelorismo Ipotonia, diastasi dei retti Deficit intellettivo e del linguaggio

119
Imprinting Figure 1. Imprint establishment and propagation during gametogenesis and development. The paternal allele (dashed line) is imprinted and the maternal allele is expressed (solid line). The "imprint mark" (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both "imprint marks" and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate "imprint marks" are reestablished for the next generation Am J Pathol 1999 Mar;154(3):635-47 Genomic Imprinting: Implications for Human Disease J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*
 is imprinted and the maternal allele is expressed (solid line). The imprint mark (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both imprint marks and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate imprint marks are reestablished for the next generation. Am J Pathol 1999 Mar;154(3): Genomic Imprinting: Implications for Human Disease. J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*")
120
Imprinting Nelle cellule germinali primordiali l’imprinting viene cancellato del tutto e il DNA è demetilato Successivamente nella linea germinale maschile si determina un pattern di imprinting che in alcuni loci è complementare a quello della linea germinale femminile I cromosomi su cui avviene l’imprinting (7, 11, 15) manterranno questo pattern e lo riprodurranno ad ogni mitosi Si potranno sempre distinguere l’espressione genica del cromosoma materno e paterno Figure 1. Imprint establishment and propagation during gametogenesis and development. The paternal allele (dashed line) is imprinted and the maternal allele is expressed (solid line). The "imprint mark" (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both "imprint marks" and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate "imprint marks" are reestablished for the next generation Am J Pathol 1999 Mar;154(3):635-47 Genomic Imprinting: Implications for Human Disease J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*
 manterranno questo pattern e lo riprodurranno ad ogni mitosi. Si potranno sempre distinguere l’espressione genica del cromosoma materno e paterno. Figure 1. Imprint establishment and propagation during gametogenesis and development. The paternal allele (dashed line) is imprinted and the maternal allele is expressed (solid line). The imprint mark (black box) represents a parental-specific methylation established during gametogenesis. A: The maternal and paternal genomes have different imprint patterns following fertilization. B: Both imprint marks and imprint reading are maintained during somatic cell division. C: The parental specific imprints are erased in the primordial germ cells. D: The appropriate imprint marks are reestablished for the next generation. Am J Pathol 1999 Mar;154(3): Genomic Imprinting: Implications for Human Disease. J. Greg Falls* , David J. Pulford* , Andrew A. Wylie* and Randy L. Jirtle*")
121
Disomia uniparentale Due copie dello stesso cromosoma sono ereditate dallo stesso genitore Spesso questo avviene attraverso un fenomeno transitorio di trisomia, seguito dalla perdita del cromosoma singolo e mantenimento del cromosoma doppio

122
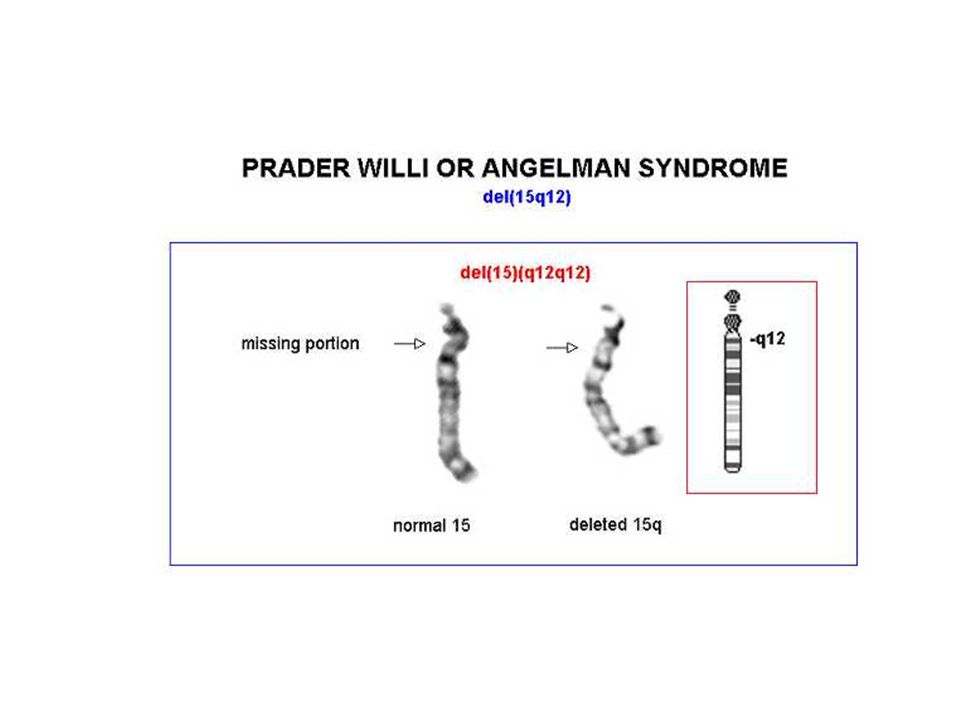
Angelman 70% dei casi delezione della regione cromosomica 15q11-q13, che è soggetta al fenomeno dell'imprinting del cromosoma paterno Il gene materno (l'unico espresso) può essere alterato con 4 meccanismi noti: delezione disomia uniparentale paterna difetti nell'imprinting mutazioni a carico del gene UBE3A (ubiquitin ligasi) La diagnosi è clinica e il difetto genetico non si identifica nel 20% dei casi
 può essere alterato con 4 meccanismi noti: delezione. disomia uniparentale paterna. difetti nell imprinting. mutazioni a carico del gene UBE3A (ubiquitin ligasi) La diagnosi è clinica e il difetto genetico non si identifica nel 20% dei casi.")
124
Angelman "happy puppet syndrome" si può identificare in Cucciolo (Dopey) "addormentato", il più giovane dei nani che non ha mai imparato a parlare ritardo mentale con assenza del linguaggio, difficoltà nell'equilibrio, eccessivo buon umore

125
Angelman L'incidenza è 1/20.000 nati
crisi epilettiche e comunque alterazioni dell'EEG e microcefalia relativa

126
Prader-Willi iperfagia>obesità eccessiva assunzione di liquidi reazioni abnormi ai sedativi acromicria, criptorchidismo insensibilità al dolore, lesioni cutanee sbalzi di umore

127
Prader-Willi 1/15.000

128
Nomenclatura delle delezioni
Le delezioni sono designate con la sigla del che segue i numeri dei nucleotidi a monte e a valle della delezione separatida un segno _ 82_83del (o 82_83delTG) indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC
 indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC.")
129
Cosa sono le distrofie muscolari?
Malattie degenerative progressive Variazione dello spessore delle miofibrille con forti cambiamenti nella istologia del muscolo indebolimento e degenerazione del tessuto muscolare in fibroso e adiposo aree di necrosi con processi infiammatori

130
Duchenne Becker cingoli Emery- Dreifuss distale facio-scapolo- omerale oculo- faringea

131
Distrofia muscolare Duchenne/Becker
DMD Duchenne - 1/3,500 maschi Insorgenza -- Infanzia - tra 2 e 6 anni Sintomi – Debolezza generalizzata e danno muscolare prima agli arti e al tronco, polpacci ingrossati Progressione – Lenta ma inesorabile. Colpisce tutti i muscoli volontari. Sopravvivenza fino a anni BMD Becker - 1/10,000 maschi Insorgenza – Adolescenza o dopo Sintomi – Identici alla DMD ma più attenuati. Vi è coinvolgimento cardiaco significativo Progressione – Più lenta e più variabile della distrofia di Duchenne con buona aspettativa di vita

132
Le delezioni intrageniche del gene della distrofina mandano fuori cornice la lettura delle triplette quando gli esoni cancellati contenevano un numero di nucleotidi che non è multiplo esatto di tre (1,2,4,5,7,8,10,11 ecc). Questo causa la distrofia di Duchenne.

134
Le delezioni intrageniche che non alterano la cornice di lettura portano alla distrofia muscolare di Becker o ad un apparente buona salute. Forniscono informazioni per preparare delle microdistrofine per la terapia genica

135
Nomenclatura delle delezioni
Le delezioni sono designate con la sigla del che segue i numeri dei nucleotidi a monte e a valle della delezione separatida un segno _ 82_83del (o 82_83delTG) indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC
 indica una delezione di TG nella sequenza ACTTTGTGCC (dove A è il nucleotide 76) che diventa ACTTTGCC.")
136
MLPA probes Each MLPA probe consists of two oligonucleotides, one synthetic and one M13-derived single-stranded DNA fragment .For each probe there is a target specific sequence that can be ligated when correctly hybridized to its target. All probes have the same PCR primer sequences at their ends. The non-hybridizing stuffer sequence of each probe has a different length and sequence enabling separation by electrophoresis

137
Hybridization The MLPA probemix is added to denatured genomic DNA
The two parts of each probe hybridise to adjacent target sequences The MLPA probe mix is added and allowed to hybridize to their respective target overnight By hybridization of probes to the target sequences, followed by a ligation reaction a copy is made of each target sequence present in the sample.

138
ligation 3. Probes are ligated by a thermostable ligase
The MLPA probe mix is added and allowed to hybridize to their respective target overnight By hybridization of probes to the target sequences, followed by a ligation reaction a copy is made of each target sequence present in the sample.

139
PCR amplification A universal primer pair is used to amplify all ligated probes The PCR product of each probe has a unique length ( bp) These ligated probe molecules are amplified in the multiplex PCR MLPA enables multiplex PCR reactions in which all specific sequences are amplified simultaneously with preserved copy numbers. This PCR reaction is very robust since only one pair of PCR primers is used for amplification of all fragments. Amplification products range in size from 130 – 490 bp and are analyzed by sequence type electrophoresis.

140
separation and quantification by capillary electrophoresis
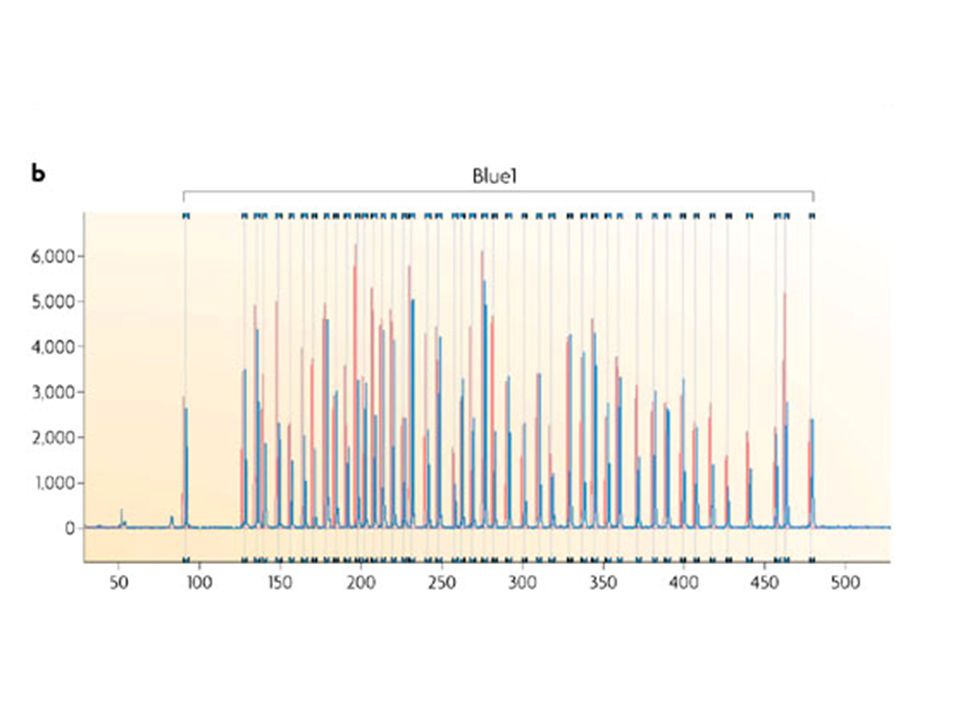
Each peak is the amplification product of a specific probe. Samples are compared to a control sample. A difference in relative peak height or peak area indicates a copy number change of the probe target sequence

143
detection of Chr X copy number
Triple X Female Male 283 bp 346 bp

144
Ligation of the two probe oligonucleotides Amplification product
MLPA discriminates sequences that differ in only a single nucleotide and can be used to detect known mutations Mismatch Perfect match Mismatch at the probe ligation site No ligation, no amplification product Ligation of the two probe oligonucleotides Amplification product

145
MS-MLPA M Methylated Target Unmethylated Target
Denaturation and Multiplex probe hybridization M Ligation and Digestion with methylation sensitive endonucleases M Hha1 Only undigested (methylated) and ligated probes are exponentially amplified
 and ligated probes are exponentially amplified.")
146
La tecnica del CGH (comparative genomic hybridization) permette l’individuazione di sequenze delete o duplicate nel genoma da testare (red) mediante il confronto con un genoma di riferimento (green). Sono preparate due sonde fluorescenti di colore diverso che ibridano contemporaneamente sui cromosomi. Se in una regione cromosomica prevale il colore (green) relativo al genoma di controllo questo significa che il genoma da testare (red) ha una delezione in quella regione
 relativo al genoma di controllo questo significa che il genoma da testare (red) ha una delezione in quella regione.")
Presentazioni simili