Scaricare la presentazione
1
oncogeni oncosoppressori
La trasformazione neoplastica è un processo che comprende una serie di mutazioni a carico di geni codificanti proteine essenziali nella proliferazione oncogeni La trasformazione neoplastica è un processo che comprende una serie di mutazioni a carico di geni codificanti proteine essenziali nella proliferazione. Semplificando, possiamo dire che il difetto base della cellula neoplastica, sia lo squilibrio tra segnali proliferativi (evidenziati in verde nella diapositiva) e quelli antiproliferativi (evidenziati in rosso), squilibrio che ha, come risultato netto, un aumento della popolazione cellulare tumorale. Il principale “relè’” su cui convergono tali stimoli è il ciclo cellulare, la cui regolazione è specificatamente alterata in un gran numero di neoplasie. Le lesioni responsabili dell'insorgenza dei tumori sono riconducibili a due ben definiti gruppi di geni, i protooncogeni e i geni oncosoppressori. In condizioni fisiologiche, le proteine codificate da questi geni partecipano al normale controllo della proliferazione cellulare in risposta agli stimoli extracellulari. I prodotti dei protooncogeni realizzano la trasmissione dei segnali proliferativi dalla superficie della cellula al suo nucleo: qui inducono l'evento chiave della replicazione della cellula, cioè la duplicazione del suo patrimonio genetico. I prodotti dei geni oncosoppressori, al contrario dei precedenti, trasmettono invece segnali inibitori, che agiscono con segno opposto sulla proliferazione cellulare. Nelle cellule neoplastiche, le lesioni genetiche riscontrate a carico dei protooncogeni sono mutazioni che comportano un'aumentata o incontrollata attività del prodotto proteico, cioè l'attivazione dell' oncogene, o entrambe queste caratteristiche. Di conseguenza, la proliferazione cellulare è iperstimolata ed eventualmente autonoma dai segnali extracellulari. Al contrario, le lesioni dei geni oncosoppressori, responsabili dell'insorgenza dei tumori, sono mutazioni inattivanti: queste, infatti, provocano la perdita di un freno alla divisione cellulare (Bishop, 1991). L'accumulo di un numero di mutazioni sufficienti per la trasformazione di una cellula normale in una cancerosa invasiva richiede, in genere, molti anni: questo giustifica un aumento dell'incidenza di tumori maligni proporzionale all'età. Inoltre, ciò significa che vi è teoricamente molto tempo per accorgersi di una neoplasia in fase iniziale e di estirparla prima che diventi maligna e letale. Tuttavia, gli stadi avanzati della trasformazione neoplastica evolvono molto rapidamente: le lesioni tendono ad accumularsi con una cinetica esponenziale, che non può essere giustificata dal semplice tasso di mutazione spontanea. Il clone neoplastico manifesta cioè una 'instabilità genetica': la responsabilità di questo fenomeno è attribuibile soprattutto alla lesione di geni che non controllano direttamente la proliferazione cellulare. l prodotti di questi geni, invece, sono preposti ai meccanismi di controllo e riparazione delle lesioni del DNA stesso, che si verificano soprattutto durante la delicata fase di replicazione del materiale genetico. La perdita dei meccanismi di difesa contro il danno genetico fa aumentare grandemente l'insorgenza di mutazioni che accelerano la selezione di cloni tumorali sempre più aggressivi (Kinzler e Vogelstein, 1996). oncosoppressori
 e quelli antiproliferativi (evidenziati in rosso), squilibrio che ha, come risultato netto, un aumento della popolazione cellulare tumorale. Il principale relè’ su cui convergono tali stimoli è il ciclo cellulare, la cui regolazione è specificatamente alterata in un gran numero di neoplasie. Le lesioni responsabili dell insorgenza dei tumori sono riconducibili a due ben definiti gruppi di geni, i protooncogeni e i geni oncosoppressori. In condizioni fisiologiche, le proteine codificate da questi geni partecipano al normale controllo della proliferazione cellulare in risposta agli stimoli extracellulari. I prodotti dei protooncogeni realizzano la trasmissione dei segnali proliferativi dalla superficie della cellula al suo nucleo: qui inducono l evento chiave della replicazione della cellula, cioè la duplicazione del suo patrimonio genetico. I prodotti dei geni oncosoppressori, al contrario dei precedenti, trasmettono invece segnali inibitori, che agiscono con segno opposto sulla proliferazione cellulare. Nelle cellule neoplastiche, le lesioni genetiche riscontrate a carico dei protooncogeni sono mutazioni che comportano un aumentata o incontrollata attività del prodotto proteico, cioè l attivazione dell oncogene, o entrambe queste caratteristiche. Di conseguenza, la proliferazione cellulare è iperstimolata ed eventualmente autonoma dai segnali extracellulari. Al contrario, le lesioni dei geni oncosoppressori, responsabili dell insorgenza dei tumori, sono mutazioni inattivanti: queste, infatti, provocano la perdita di un freno alla divisione cellulare (Bishop, 1991). L accumulo di un numero di mutazioni sufficienti per la trasformazione di una cellula normale in una cancerosa invasiva richiede, in genere, molti anni: questo giustifica un aumento dell incidenza di tumori maligni proporzionale all età. Inoltre, ciò significa che vi è teoricamente molto tempo per accorgersi di una neoplasia in fase iniziale e di estirparla prima che diventi maligna e letale. Tuttavia, gli stadi avanzati della trasformazione neoplastica evolvono molto rapidamente: le lesioni tendono ad accumularsi con una cinetica esponenziale, che non può essere giustificata dal semplice tasso di mutazione spontanea. Il clone neoplastico manifesta cioè una instabilità genetica : la responsabilità di questo fenomeno è attribuibile soprattutto alla lesione di geni che non controllano direttamente la proliferazione cellulare. l prodotti di questi geni, invece, sono preposti ai meccanismi di controllo e riparazione delle lesioni del DNA stesso, che si verificano soprattutto durante la delicata fase di replicazione del materiale genetico. La perdita dei meccanismi di difesa contro il danno genetico fa aumentare grandemente l insorgenza di mutazioni che accelerano la selezione di cloni tumorali sempre più aggressivi (Kinzler e Vogelstein, 1996). oncosoppressori.")
2
(CHROMOSOME REPLICATION) S-PHASE
CELL DIFFERENTIATION CELL DIVISION Cell cycle and cytotoxic opportunities G1 PERIOD CELL LIFE CYCLE G2 PERIOD 2. Cytotoxic Agents: Cell Cycle and Cytotoxic Opportunities Uncontrolled cell cycling and differentiation are hallmarks of cancer pathology. Rapidly-dividing cells, such as many cancerous cells, are susceptible to interventions that disrupt the molecular events of the various phases of the cell cycle. For example, the S phase, during which cellular DNA is synthesized in preparation for cell division, offers important cytotoxic opportunities due to the cell’s requirement for purines and pyrimidines with which to assemble the macromolecular nucleic acids. The process of cell division is another phase of the cell cycle that is particularly amenable to interruption with chemotherapy. TIME Farmaci “selettivamente” antiproliferativi (CHROMOSOME REPLICATION) S-PHASE
 S-PHASE.")
3
Agenti citotossici Alkylating Agents Anti- Metabolites Mitotic Inhibitors Antibiotics Others Busulfan Cytosine Etoposide Bleomycin L-asparaginase Carmustine Arabinoside Teniposide Dactinomycin Hydroxyurea Chlorambucil Floxuridine Vinblastine Daunorubicin Procarbazine Cisplatin Fluorouracil Vincristine Doxorubicin Cyclophosphamide Mercaptopurine Vindesine Mitomycin-c Ifosfamide Methotrexate Taxoids Mitoxantrone Melphalan Plicamycin 3. Principles of Chemotherapy: Classification of Cytotoxic Agents Twenty anticancer agents account for over 95% of all prescriptions in chemotherapy. They are divided into five major classifications.

4
Effetti collaterali indotti dagli agenti citotossici
Mucositis Nausea/vomiting Diarrhea Cystitis Sterility Myalgia Neuropathy Alopecia Pulmonary fibrosis Cardiotoxicity Local reaction Renal failure Myelosuppression Phlebitis 10. Principles of Chemotherapy: Side Effects of Chemotherapy There are multiple side effects of chemotherapy. Some are common, such as alopecia, neutropenia. Some are rare such as cardiotoxicity. The side effects are generally the consequence of the cytotoxic effect of chemotherapy on normal cells, however they can also be related to the direct toxicity of the drug.

5
Sviluppo clinico degli agenti citotossici
Tipico effetto citotossico OBD >MTD Effetto Tossicità Dose Target Effetto antitumorale In traditional clinical trials the recommended dose is usually the maximum dose that is tolerated by patients (the maximum tolerated dose [MTD]) due to the toxicity of standard cytotoxic agents. This dose is not necessarily the dose at which maximum efficacy is observed. For novel targeted agents that are cytostatic, antitumor activity may be observed well below the MTD and for these agents the optimal biologic dose (OBD) may be used.1 As such, the OBD generates maximum efficacy with minimum toxicity. Reference Rowinsky EK. Drugs 2000; 60 (Suppl 1): 1-14. Figure reproduced with permission from: Rowinsky EK. The pursuit of optimal outcomes in cancer therapy in a new age of rationally designed target-based anticancer agents. Drugs 2000; 60 (Suppl 1): 1-14. MTD OBD OBD, dose biologica ottimale; MTD, massima dose tollerata Rowinsky 2000
 due to the toxicity of standard cytotoxic agents. This dose is not necessarily the dose at which maximum efficacy is observed. For novel targeted agents that are cytostatic, antitumor activity may be observed well below the MTD and for these agents the optimal biologic dose (OBD) may be used.1 As such, the OBD generates maximum efficacy with minimum toxicity. Reference. Rowinsky EK. Drugs 2000; 60 (Suppl 1): Figure reproduced with permission from: Rowinsky EK. The pursuit of optimal outcomes in cancer therapy in a new age of rationally designed target-based anticancer agents. Drugs 2000; 60 (Suppl 1): MTD. OBD. OBD, dose biologica ottimale; MTD, massima dose tollerata Rowinsky")
6
Efficacia della chemioterapia nelle neoplasie del colon-retto in fase metastatica

7
Bersagli Molecolari General target Specific target Agent or approach
Signal transduction Growth factor receptors HER1 (EGFR) HER2 Bcr - Abl Ras Raf Tarceva TM Iressa Herceptin Glivec Farnesyl transferase inhibitors Antisense oligonucleotides Angiogenesis and metastasi s VEGFR2 VEGF Matrix metalloproteinases Integrins SU5416 rhuMAb (Avastin ) Neovastat Vitaxin Tumour suppressor gene p53 p16 Gene therapy Cell cycle control Cyclin dependent kinases Flavopiridol Cetuximab
 HER2. Bcr. - Abl. Ras. Raf. Tarceva. TM. Iressa. Herceptin. Glivec. Farnesyl transferase inhibitors. Antisense oligonucleotides. Angiogenesis and. metastasi. s. VEGFR2. VEGF. Matrix metalloproteinases. Integrins. SU5416. rhuMAb (Avastin. ) Neovastat. Vitaxin. Tumour suppressor. gene. p53. p16. Gene therapy. Cell cycle control. Cyclin. dependent. kinases. Flavopiridol. Cetuximab.")
8
Sviluppo clinico degli agenti biologici a “bersaglio specifico”
Farmaci cytotossici OBD >MTD Agenti biologici OBD <MTD Effetto Effetto Tossicità Tossicità Effetto antitumorale Dose Target Effetto antitumorale Dose Target In traditional clinical trials the recommended dose is usually the maximum dose that is tolerated by patients (the maximum tolerated dose [MTD]) due to the toxicity of standard cytotoxic agents. This dose is not necessarily the dose at which maximum efficacy is observed. For novel targeted agents that are cytostatic, antitumor activity may be observed well below the MTD and for these agents the optimal biologic dose (OBD) may be used.1 As such, the OBD generates maximum efficacy with minimum toxicity. Reference Rowinsky EK. Drugs 2000; 60 (Suppl 1): 1-14. Figure reproduced with permission from: Rowinsky EK. The pursuit of optimal outcomes in cancer therapy in a new age of rationally designed target-based anticancer agents. Drugs 2000; 60 (Suppl 1): 1-14. Dose Dose MTD OBD OBD MTD OBD, dose biologica ottimale; MTD, massima dose tollerata Rowinsky 2000
 due to the toxicity of standard cytotoxic agents. This dose is not necessarily the dose at which maximum efficacy is observed. For novel targeted agents that are cytostatic, antitumor activity may be observed well below the MTD and for these agents the optimal biologic dose (OBD) may be used.1 As such, the OBD generates maximum efficacy with minimum toxicity. Reference. Rowinsky EK. Drugs 2000; 60 (Suppl 1): Figure reproduced with permission from: Rowinsky EK. The pursuit of optimal outcomes in cancer therapy in a new age of rationally designed target-based anticancer agents. Drugs 2000; 60 (Suppl 1): Dose. Dose. MTD. OBD. OBD. MTD. OBD, dose biologica ottimale; MTD, massima dose tollerata Rowinsky")
9
Un esempio paradigmatico di terapia molecolare bersaglio-specifica
“il recettore per l’EGF”

10
No specific ligands - often acts as dimer partner
The EGFR (erbB) family EGF TGF- Amphiregulin -cellulin HB-EGF Epiregulin Ligands No specific ligands - often acts as dimer partner NRG2 NRG3 Heregulins -cellulin Heregulins Extracellular Cysteine-rich Receptor domain Membrane Intracellular Il recettore per il fattore di crescita epidermoidale (o EGFR) fa parte di una famiglia composta da quattro membri strutturalmente correlati tra di loro. Sono glicoproteine transmebrana, caratterizzate dalla presenza di un dominio extracellulatre, deputato a riconoscere specifici ligandi, da un dominio transmebrana e da uno intracitoplasmatico, con attività tirosin-chinasica. Essi sono: EGFR (HER1 or erbB1),erbB2 (HER2/neu), erbB3 (HER3), and erbB4 (HER4). Tali recettori hanno la capacità di legare diversi ligandi. L’EGF e il TGF alfa sono i più noti, rappresentando proteine importanti nel mantenimento della crescita neoplastica, e si legano al dominio extracellulare di ErbB1. A differenza degli altri recettori, per ErbB2 non sono stati definiti ligandi specifici, mentre ErbB3 no ha attività tirosin chinasica e 4 legano hereguline e neuroreguline Tyrosine kinase domain K K K K erbB1 HER1 EGFR erbB2 HER2 neu erbB3 HER3 erbB4 HER4 Wells 1999
 family. EGF. TGF- Amphiregulin. -cellulin. HB-EGF. Epiregulin. Ligands. No specific ligands - often acts as dimer partner. NRG2. NRG3. Heregulins. -cellulin. Heregulins. Extracellular. Cysteine-rich. Receptor domain. Membrane. Intracellular. Il recettore per il fattore di crescita epidermoidale (o EGFR) fa parte di una famiglia composta da quattro membri strutturalmente correlati tra di loro. Sono glicoproteine transmebrana, caratterizzate dalla presenza di un dominio extracellulatre, deputato a riconoscere specifici ligandi, da un dominio transmebrana e da uno intracitoplasmatico, con attività tirosin-chinasica. Essi sono: EGFR (HER1 or erbB1),erbB2 (HER2/neu), erbB3 (HER3), and erbB4 (HER4). Tali recettori hanno la capacità di legare diversi ligandi. L’EGF e il TGF alfa sono i più noti, rappresentando proteine importanti nel mantenimento della crescita neoplastica, e si legano al dominio extracellulare di ErbB1. A differenza degli altri recettori, per ErbB2 non sono stati definiti ligandi specifici, mentre ErbB3 no ha attività tirosin chinasica e 4 legano hereguline e neuroreguline. Tyrosine kinase. domain. K. K. K. K. erbB1 HER1. EGFR. erbB2. HER2. neu. erbB3. HER3. erbB4. HER4. Wells")
11
X Extracellulare Membrana Intracellulare K K K K K K P P P P P P
Ligando EGF TGF- Amphiregulin -cellulin HB-EGF Epiregulin X Extracellulare Membrana Intracellulare dominio tirosin chinasico EGFR homodimerization EGFR heterodimerization with erbB2 or erbB3 receptors erbB4 heterodimerization with erbB2.1 Dimer formation will dictate the resultant signaling cascade.1 Reference Wells A. Int J Biochem Cell Biol 1999; 31: L’ingaggio del ligando con il dominio extracellulare del recettore, ne induce la rapida dimerizzazione. Tale evento può avvenire sia con recettori omologhi (omodmerizzazione) che con altri membri della famiglia (etero dimerizzazione). Il tipo di associazione (omodimerizzazione o eterodimerizzazione) dipendono dal tipo di tessuto, dallo stato funzionale e dalla diversa distribuzione spaziale e/o temporale dei doversi recettori. Nessun ligando è in grado d’intereagire con ErbB2, ma ERBB2 è il recettore che con maggiore frequenza dimerizza con gli altri membri dopo l’attivazione dei recettori della famiglia Erb. ErbB3 ha ridotta attività kinasica e acquisisce tale capacità solo dimerizzandosi con altri membri della famiglia (eterodimerizzazione -ERBB2). Per esempio, in presenza di un’ elevata espressione di ErbB2, si fprmeranno preferibilmente eterodimeri ERbB1-ErbB2 e quest’evento, come vedremo in seguito, è un evento in grado di conferire aggressività al tumore mammario, e , probabilmente, anche ad altre neoplasie solide K K K K K K P P P P P P erbB1 HER1 EGFR erbB2 HER2neu omodimero HER1/HER1 eterodimero HER1/HER2neu Wells 1999
 che con altri membri della famiglia (etero dimerizzazione). Il tipo di associazione (omodimerizzazione o eterodimerizzazione) dipendono dal tipo di tessuto, dallo stato funzionale e dalla diversa distribuzione spaziale e/o temporale dei doversi recettori. Nessun ligando è in grado d’intereagire con ErbB2, ma ERBB2 è il recettore che con maggiore frequenza dimerizza con gli altri membri dopo l’attivazione dei recettori della famiglia Erb. ErbB3 ha ridotta attività kinasica e acquisisce tale capacità solo dimerizzandosi con altri membri della famiglia (eterodimerizzazione -ERBB2). Per esempio, in presenza di un’ elevata espressione di ErbB2, si fprmeranno preferibilmente eterodimeri ERbB1-ErbB2 e quest’evento, come vedremo in seguito, è un evento in grado di conferire aggressività al tumore mammario, e , probabilmente, anche ad altre neoplasie solide. K. K. K. K. K. K. P. P. P. P. P. P. erbB1 HER1. EGFR. erbB2. HER2neu. omodimero HER1/HER1. eterodimero. HER1/HER2neu. Wells")
12
La formazione di omo o etero-dimeri porta all’attivazione del dominio kinasico intracellulare; tale attivazione induce l’autofosforilazione di specifici residui tirosinici presenti al livello della coda intracitoplasmatica. I residui fosforilati possono essere considerati alla stregua di siti d’ancoraggio e di fosforilazione per specifiche proteine di segnale intracellulare. In questa diapositiva, sono shematicamente rappresentati i principali siti di autofosforilazione di EGFR, ErbB2 e ErbB3 e delle proteine di segnale associate. Nonostante una sostanziale rindondanza in termini di molecole reclutate, è osservabile una certa specificità. Es neoplasie che esprimono EGFR con mutazione a carico del dominio kinasico, attivano preferenzialmente PI3K–AKT e i pathways di STAT. L’EGFR, pur non avendo consensus sequence per la proteina adttatrice p85 di PI3K, è in grado di attivare tale pathway tramite GAB1, che lega GRB2. STAT viene attivato indirettamente, attraverso il resuduo in tyrosine-1068 e (Ref. 137). Ancora, ERBB2 modula la via di MAPK attraverso GRB2, SHC, downstream of kinase related (DOK-R)138, CRK e la phospholipase C (PLC ). Pur non evendo intrinseca attività kinasica, ERBB3 viene attivato dal legame con le neureguline, dimerezzinandosi avidamente con ErbB2 7. ERBB3 contiene sei docking sites per p85, subunità adattatrice di PI3K e modula molto eficientemente tale pathaway AR, amphiregulin; BTC, betacellulin; EPR, epiregulin; HB-EGF, heparin-binding EGF; NRGs, neuregulins; TGF , transforming growth factor- Nature Rev .Vol 5:341, 2005
138, CRK e la phospholipase C (PLC ). Pur non evendo intrinseca attività kinasica, ERBB3 viene attivato dal legame con le neureguline, dimerezzinandosi avidamente con ErbB2 7. ERBB3 contiene sei docking sites per p85, subunità adattatrice di PI3K e modula molto eficientemente tale pathaway. AR, amphiregulin; BTC, betacellulin; EPR, epiregulin; HB-EGF, heparin-binding EGF; NRGs, neuregulins; TGF , transforming growth factor- Nature Rev .Vol 5:341,")
13
Ruolo fisiologico dei recettori della famiglia Erb
EGFR -/- Alterazioni sviluppo epitelio intestinale, epitelio polmonare, cute Neurodegenerazione talamica e corticale ErbB2-/- Alterazioni sviluppo cardiaco “heart-restricted ErbB2-inactivation” Cardiopatia dilatativa Qual è il ruolo fisiologico di tali recettori? Topi KO per l’ EGFR muoiono durante la prima settimana di vita per insufficienz respiratoria. Inoltre, manifestano alterazioni a livello degli epiteli gastrointestinali e cutaneo (pelle sottile, alterazioni follicolari) e neurodegenerazione nelle regioni corticali e del talamo. Tali osservazioni possono spiegare gli effetti collaterali registrati indotti dal trattamento con agenti (MoAb, TKI) in grado di bloccare l’attività dell’EGFR: rash, follicolite,diarrea. Anche se infrequente (1% ), l’interstiziopatia polmonare è stata associata al trattamento con gefitinib in pz. con NSCLC; soggetti con comorbidità polmonari, come la fibrosi polmonare idiopatica, sembrano essere sensibilmente più a rischio di sviluppare tale effetto collaterale. ERBB2 ha un ruolo essenziale nello sviluppo cardiaco Embrioni KO per il gene ErbB2 muoiono per mancato sviluppo ventricolare. La soppressione selettiva dell’espressione di ErbB2 a livello dei cardiomiociti induce lo sviluppo di una miocardiopatia dilatativa, e una maggiore sensibilità alle antracicline; ciò spiega l’ insorgenza di tale patologia in pazienti trattati con anticorpi anti-ErbB2, soprattutto quando associato alle antracicline. Alterazioni dello sviluppo cardiaco sovrapponibili sono indotte anche dall’assenza di ErbB4 o del suo ligando, neuregulina-1, dimostrando l’mportanza del dimero ErbB2-4 nello sviluppo cardiaco. ErbB4-/- Alterazioni sviluppo cardiaco/neurale Neuregulina 1-/- Alterazioni sviluppo cardiaco
 e neurodegenerazione nelle regioni corticali e del talamo. Tali osservazioni possono spiegare gli effetti collaterali registrati indotti dal trattamento con agenti (MoAb, TKI) in grado di bloccare l’attività dell’EGFR: rash, follicolite,diarrea. Anche se infrequente (1% ), l’interstiziopatia polmonare è stata associata al trattamento con gefitinib in pz. con NSCLC; soggetti con comorbidità polmonari, come la fibrosi polmonare idiopatica, sembrano essere sensibilmente più a rischio di sviluppare tale effetto collaterale. ERBB2 ha un ruolo essenziale nello sviluppo cardiaco Embrioni KO per il gene ErbB2 muoiono per mancato sviluppo ventricolare. La soppressione selettiva dell’espressione di ErbB2 a livello dei cardiomiociti induce lo sviluppo di una miocardiopatia dilatativa, e una maggiore sensibilità alle antracicline; ciò spiega l’ insorgenza di tale patologia in pazienti trattati con anticorpi anti-ErbB2, soprattutto quando associato alle antracicline. Alterazioni dello sviluppo cardiaco sovrapponibili sono indotte anche dall’assenza di ErbB4 o del suo ligando, neuregulina-1, dimostrando l’mportanza del dimero ErbB2-4 nello sviluppo cardiaco. ErbB4-/- Alterazioni sviluppo cardiaco/neurale. Neuregulina 1-/- Alterazioni sviluppo cardiaco.")
14
Espressione dell’EGFR nei tumori umani
Tumore Espressione Prognosi Rischio di metastasi Colon-retto 25-77% avversa aumentato Testa-collo 95-100% DFS, OAS Pancreas 30-89% OAS NSCLC 40-80% OAS Rene 50-90% Vescica 30-48% Ovaio 35-70% L’iperespressione del recettore per l’EGF è un evento comune nei tumori solidi. L’espressione dell’ EGFR è molto comune(es., dal 35% al 70% nei tumori ovarici e dal 25% al 77% nelle neoplasie del colon-retto ed è frequentemente correlato con un peggioramento della prognosi e ad una maggiore aggressività. In alcuni casi, inoltre, l’espressione dell’EGFR è direttamente correlta all’incremento della resistenza a chemioterapici e alla RT.31,32 Per il suo ruolo nella promozione della crescita tumorale appare , quindi, razionale l’inibizione selettiva del segnale mediato dall’attivazione dell’EGFR. Studi preclinici hanno inequivocabilmente dimostrato che il blocco dell’EGFR è in grado d’inibire la crescita di neoplasie EGFR positive.41,42

15
EGFR signal transduction in tumor cells
Ligand EGFR K K EGFR-TK PI3-K GRB2 pY pY SOS pY STAT3 RAS RAF PTEN AKT MEK Nello specifico, l’attivazione dell’EGFR genera una cascata di segnali intracellulari in grado di promuovere la proliferazione cellulare, attraverso l’attivazione di MAPK, la sopravvivenza cellulare attraverso l’attivazione di AKT e la neoangiogenesi. Non stupisce, quindi, che l’iperespressione dell’EGFR possa aumentare l’indice proliferativo e le potenzialità metastatiche di una neoplasia References Wells A. Int J Biochem Cell Biol 1999; 31: Woodburn JR. Pharmacol Ther 1999; 82: Hanahan D, Weinberg RA. Cell 2000; 100: Akimoto T et al. Clin Cancer Res 1999; 5: Raymond E et al. Drugs 2000; 60 (Suppl 1): Balaban N et al. Biochim Biophys Acta 1996; 1314: Gene transcription Cell cycle progression MAPK P P DNA Myc Cyclin D1 Proliferation / maturation JunFos Cyclin D1 Survival (anti-apoptosis) Myc Chemotherapy / radiotherapy resistance Metastasis Angiogenesis
: Balaban N et al. Biochim Biophys Acta 1996; 1314: Gene transcription. Cell cycle progression. MAPK. P P. DNA. Myc. Cyclin D1. Proliferation / maturation. JunFos. Cyclin D1. Survival (anti-apoptosis) Myc. Chemotherapy / radiotherapy resistance. Metastasis. Angiogenesis.")
16
Perchè inibire le funzioni dell’EGFR?
L’attivazione dell’EGFR promuove la crescita e la sopravvivenza delle cellule tumorali L’espressione o l’iperespressione è un evento comune in numerosi tipi di neoplasie solide. Tale espresssione è generalmente associata ad un peggioramento della prognosi Pertanto, l’inibizione funzionale dell’EGFR rappresenta un approccio razionale nel trattamento delle neoplasie umane As the EGFR is part of a signal transduction pathway that controls cell proliferation in normal tissues, high expression or hyperactivation of EGFR (eg by an excess of ligand or a reduction in turnover) can result in increased signaling that leads to tumor cell survival and proliferation. As the EGFR is expressed or highly expressed in many solid tumors, inhibition of the EGFR signaling pathway forms a rational target for anticancer therapy.
 can result in increased signaling that leads to tumor cell survival and proliferation. As the EGFR is expressed or highly expressed in many solid tumors, inhibition of the EGFR signaling pathway forms a rational target for anticancer therapy.")
17
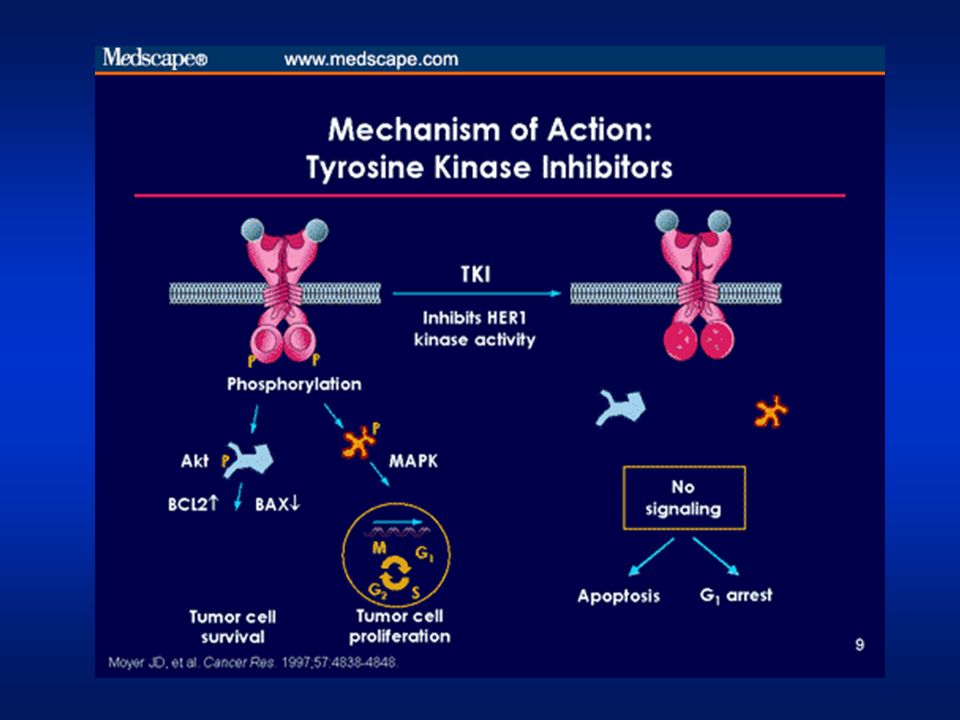
Strategies to inhibit EGFR signaling
Immune effector cell Anti-ligand mAbs Bispecific Abs Ligand / toxin conjugate Anti-EGFR mAbs (eg cetuximab) Nucleus During the complex processes following ligand binding (eg signal transduction and altered gene expression) there are many opportunities for therapeutic intervention, which include: Anticorpi monoclonali (mAbs) che si legano al recettore prevenendo il legame con il ligando (cetuximab) Inibitori dell’attività tirosina kinasica del recettore (EGFR-TKIs) (gefitinib, erlotinib) Anticorpi bi-speciici in grado di legarsi contemporaneamente all’EGFR e una cellula immunitaria effettrice in grado di uccidere la cellula tumorale Anticorpi anti-ligando Anticorpi anti EGFR coniugato con tossine o con inibitori specifici (such as genistein) Reference Noonberg SB, Benz CC. Drugs 2000; 59: Figure reproduced with permission from: Noonberg SB, Benz CC. Tyrosine kinase inhibitors targeted to the epidermal growth factor receptor subfamily: role as anticancer agents. Drugs 2000; 59: Y EGFR tyrosine kinase inhibitors (EGFR-TKIs) (gefitinib,erlotinib) Adapted from Noonberg & Benz 2000
 Nucleus. During the complex processes following ligand binding (eg signal transduction and altered gene expression) there are many opportunities for therapeutic intervention, which include: Anticorpi monoclonali (mAbs) che si legano al recettore prevenendo il legame con il ligando (cetuximab) Inibitori dell’attività tirosina kinasica del recettore (EGFR-TKIs) (gefitinib, erlotinib) Anticorpi bi-speciici in grado di legarsi contemporaneamente all’EGFR e una cellula immunitaria effettrice in grado di uccidere la cellula tumorale. Anticorpi anti-ligando. Anticorpi anti EGFR coniugato con tossine o con inibitori specifici (such as genistein) Reference. Noonberg SB, Benz CC. Drugs 2000; 59: Figure reproduced with permission from: Noonberg SB, Benz CC. Tyrosine kinase inhibitors targeted to the epidermal growth factor receptor subfamily: role as anticancer agents. Drugs 2000; 59: Y. EGFR tyrosine kinase inhibitors (EGFR-TKIs) (gefitinib,erlotinib) Adapted from Noonberg & Benz")
19
Inibitori dell’attività TK dei recettori della famiglia dell’EGF
Agente Molecole/Legame Specificità Neoplasie Fase erlotinib anilinchinazolinico reversibile HER1 TK NSCLC, HN, mammella,ovaio, prostata, pancreas, colon, glioma Appr. gefitinib anilinchinazolinico reversibile HER1 TK NSCLC, HN, mammella,ovaio, prostata, pancreas, colon, glioma Appr. EKR-569 3-cianochinolone irreversibile HER1/2 TK NSCLC, mammella, altre neoplasie con erbB alterato 1/2. Il numero di tali farmaci è in continua espansione. In questa diapositiva sono illustrati i principali inibitori dell’attività TK. Alcuni di questi, come l’erlotinib e il gefitinib sono registrati per l’utilizzo clinico. Altri sono in avanzato stadio di sperimentazione, alcuni ormai prossimi alla registrazione (lapatinib). Sono composti strutturalmente semplici alcuni con proprietà bifunzionali, in quanto capaci d’inibire l’attività TK di più di un membro della famiglia Erb CI-1033 anilinchinazolinico irreversibile HER1-4 TK NSCLC, mammella, altre neoplasie con erbB alterato 1/2. lapatinib 6-tiazolilchinazolinico reversibile HER1/2 TK Mammella, neoplasie con erbB alterato 3
. Sono composti strutturalmente semplici alcuni con proprietà bifunzionali, in quanto capaci d’inibire l’attività TK di più di un membro della famiglia Erb. CI anilinchinazolinico. irreversibile. HER1-4 TK. NSCLC, mammella, altre neoplasie con erbB alterato. 1/2. lapatinib. 6-tiazolilchinazolinico. reversibile. HER1/2 TK. Mammella, neoplasie con erbB alterato. 3.")
20
Cetuximab (C225) IgG1 MAb (chimerico)
Legame specifico all’EGFR e ai suoi eterodimeri Alta affinità per l’EGFR (1 log maggiore del ligando naturale) Emivita 114 ore (75-188) Stimola l’internalizzazione del recettore Blocca la dimerizzazione del recettore, la sua attività TK, e la trasduzione del segnale. MAbs Ligando K K Trasduzione del segnale
 Emivita 114 ore (75-188) Stimola l’internalizzazione del recettore. Blocca la dimerizzazione del recettore, la sua attività TK, e la trasduzione del segnale. MAbs. Ligando. K. K. Trasduzione. del segnale.")
21
Gefitinib blocks the growth of established xenografts
Vehicle Tumor volume (mm3) 2.0 1.6 1.2 Gefitinib blocked the growth of established A431 (vulval squamous carcinoma) human tumor xenografts.1 Tumors were 1 month old at the start of therapy and treatment with gefitinib (200 mg/kg/day) for 90 days produced rapid tumor regression in all mice.1 Following drug withdrawal, the use of vehicle alone (Days ) led to the rapid re-establishment of four out of six tumors.1 Reference Wakeling A et al. Cancer Res 2002; 62: Figure reproduced with permission from: Wakeling A et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signalling with potential for cancer therapy. Cancer Res 2002; 62: ©American Association for Cancer Research. 0.8 0.4 0.0 100 80 60 40 20 120 Wakeling et al 2002 Duration of treatment (days)
 Gefitinib blocked the growth of established A431 (vulval squamous carcinoma) human tumor xenografts.1. Tumors were 1 month old at the start of therapy and treatment with gefitinib (200 mg/kg/day) for 90 days produced rapid tumor regression in all mice.1. Following drug withdrawal, the use of vehicle alone (Days ) led to the rapid re-establishment of four out of six tumors.1. Reference. Wakeling A et al. Cancer Res 2002; 62: Figure reproduced with permission from: Wakeling A et al. ZD1839 (Iressa): an orally active inhibitor of epidermal growth factor signalling with potential for cancer therapy. Cancer Res 2002; 62: ©American Association for Cancer Research Wakeling et al Duration of treatment (days)")
22
Cetuximab inhibits the invasion of established xenografts
Tumor volume (mm3) 220 15 Locoregional invasion tissuea Control Cetuximab (0.5 mg/bi-weekly) Muscle 6/8 3/8 Vascular 5/8 1/8* Bone 5/8 1/8* Perineural 3/8 0/8* Cetuximab has also been shown to inhibit SCC-1 invasion in vivo.1 Tumors were 10 days old at the start of treatment with cetuximab (0.8 mg/bi-weekly), and invasion evaluated on Day 50.1 Invasion of SCC-1 cells into the muscle was reduced by treatment with cetuximab when compared with control treatment, although this was not significant (p=0.13).1 Treatment with cetuximab significantly inhibited the invasion of SCC-1 cells into the vascular, bone, and perineural tissues when compared to control (p<0.05 for all).1 Reference Huang S-M et al. Mol Cancer Ther 2002; 1: Table reproduced with permission from: Huang S-M et al. Molecular inhibition of angiogenesis and metastatic potential in human squamous cell carcinomas after epidermal growth factor receptor blockade. Mol Cancer Ther 2002; 1: ©American Association for Cancer Research. aNumber of mice with invasion/number of injected mice *p<0.05 Huang et al 2002
 Locoregional invasion tissuea. Control. Cetuximab (0.5 mg/bi-weekly) Muscle. 6/8. 3/8. Vascular. 5/8. 1/8* Bone. 5/8. 1/8* Perineural. 3/8. 0/8* Cetuximab has also been shown to inhibit SCC-1 invasion in vivo.1. Tumors were 10 days old at the start of treatment with cetuximab (0.8 mg/bi-weekly), and invasion evaluated on Day Invasion of SCC-1 cells into the muscle was reduced by treatment with cetuximab when compared with control treatment, although this was not significant (p=0.13).1. Treatment with cetuximab significantly inhibited the invasion of SCC-1 cells into the vascular, bone, and perineural tissues when compared to control (p<0.05 for all).1. Reference. Huang S-M et al. Mol Cancer Ther 2002; 1: Table reproduced with permission from: Huang S-M et al. Molecular inhibition of angiogenesis and metastatic potential in human squamous cell carcinomas after epidermal growth factor receptor blockade. Mol Cancer Ther 2002; 1: ©American Association for Cancer Research. aNumber of mice with invasion/number of injected mice. *p<0.05. Huang et al")
23
Cell cycle analysis by flow cytometry
SCC-13Y cells 48 hrs G1 G1 G2M G2M S S Harari PM and Huang SM Int. J. Radiation Oncology Biol. Phys., 2001

24
Effetto della combinazione C225+RT
linea cellulare SSC-1 Harari PM and Huang SM Int. J. Radiation Oncology Biol. Phys., 2001

25
Flow cytometric determination of apoptosis by DNA staining
Radiation-induced apoptosis -SCC-13Y cells- Flow cytometric determination of apoptosis by DNA staining sub-diploid population Harari PM and Huang SM Int. J. Radiation Oncology Biol. Phys., 2001

27
Un altro punto da sottolineare e l’importanza della sequenza farmacologici. Quando si utilizzano combinazioni farmacologiche comprendenti inibitori dell’attività dell’EGFR e farmaci citotossici, i risultati possono cambiare drammaticamente a seconda della schedula di somministrazione. In questa diapositiva si vede chiaramente come la sequenza erlotinib-paclitaxel sia in grado di antagonizzare gli effetti sul ciclo cellulare e sulla crescita del taxano. Da qui la raccomandazione che qualsiasi associazione dovrebbe essere testata con attenzione in modelli preclinici anche per quanto riguarda la sequenza di somministrazione

28
Fattori in grado di determinare la sensibilità all’inibizione del recettore dell’EGF
La cellula deve dipendere, per la sua sopravvivenza, dall’aumentata espressione dell’EGFR (oncogene addiction) Amplificazione genica (ErbB1 – ErbB2) Presenza di mutazioni attivanti costitutivamente il recettore (ErbB1)
 Amplificazione genica (ErbB1 – ErbB2) Presenza di mutazioni attivanti costitutivamente il recettore (ErbB1)")
29
tempo FISH Amplificazione genica (EGF-R) Vantaggio selettivo (EGF-R)
Dobbiamo tenere conto che l’attività di un farmaco a bersaglio specifico è funzione della dipendenza della cellula nei confronti del pathway inibito dallo specifico farmaco. Nel caso dell’EGFR, una popolazione cellulare può esprimere il recettore in risposta ad una situazione contingente e temporanea (es. trattamenti terapeutici, radioterapia). In altri casi, un’alterazione genetica specifica, quale l’amplificazione del gene delll’EGFR e la conseguente iperespressione del recettore, può rappresentare un vantaggio selettivo durante l’evoluzione delle neoplasia. In questo caso la popolazione selezionata è strettamente dipendente dalla presenza e dall’attività del recettore stesso. Iper-espressione temporanea (EGF-R)
. In altri casi, un’alterazione genetica specifica, quale l’amplificazione del gene delll’EGFR e la conseguente iperespressione del recettore, può rappresentare un vantaggio selettivo durante l’evoluzione delle neoplasia. In questo caso la popolazione selezionata è strettamente dipendente dalla presenza e dall’attività del recettore stesso. Iper-espressione temporanea (EGF-R)")
30
2- 4 5 - 7 8 - 12 13 -16 17 25 -28 Regione transmembrana Dominio TK
Sito ATP Dominio extracellulare Dominio regolatorio N-Lobe C-Lobe 2- 4 5 - 7 8 - 12 13 -16 17 25 -28 Anche la presenza di mutazioni a livello recettoriale, possono conferire un vantaggio selettivo. A tale proposito, sono state identificate e caratterizzate alcune mutazioni a carico del dominio TK del recettore dell’EGF, mutazioni che conferiscono attività costitutiva al recettore stesso. Alcune di queste mutazioni, riscontrate essenzialmente nelle neoplasie polmonari di tipo adenocarcinomatoso conferiscono alla cellula una spiccata sensibilità a piccole molecole ininbitorie dell’attività TK,come il gefitinib, dando conto della spiccata sensibilità a tale strategia terapeutica di tali tumori. Altre mutazioni, invece, sembrerebbero conferire resistenza al trattamento con ininbitori specifici dell’attività TK Mutazione puntiformi esone 21 (L858R) (39%) Delezione esone 19 (46%) Mutazione puntiformi esone 20 (T790M)
 (39%) Delezione esone 19 (46%) Mutazione puntiformi esone 20 (T790M)")
31
Fattori in grado di determinare la sensibilità all’inibizione del recettore dell’EGF
La cellula deve dipendere, per la sua sopravvivenza, dall’aumentata espressione dell’EGFR (oncogene addiction) Adeguati livelli d’inibizione dell’EGFR dose farmacocinetica tipo d’inibitore usato
 Adeguati livelli d’inibizione dell’EGFR. dose. farmacocinetica. tipo d’inibitore usato.")
32
L’introduzione di farmaci biologici capaci d’inibire selettivamente l’attività kinasica di un bersaglio molecolare ha radicalmente cambiato il concetto di dose efficace. Mentre con i classici farmaci abituati ad utilizzare la massima dose tollerata, per ueste molecole è considerata la dose biologicamente efficace, cioè la dose in grado d’inibire l’attività biologica del bersaglio. Tale dose, però, non sempre corrisponde alla completa inibizione del pathway innescato dalla molecola bersaglio, probabilmente per la presenza di vie di attivazione collaterali. Come si può vedere nella diapositiva, dosi crescenti dell’inibitore specifico dell’attività TK del recettore dell’EGF, erlotinib, è in grado d’inibire l’autifosforilazione dei residui tirosinici del recettore. Di contro,l’inibizione dell’attività di MAPK, uno dei bersagli principali del recettore per l’EGFR è ancora presente a dosaggi di erlotinib in grado d’inibire in modo quasi completo l’attività TK del recettore dell’EGF.

33
Fattori in grado di determinare la sensibilità all’inibizione del recettore dell’EGF
La cellula deve dipendere, per la sua sopravvivenza, dall’aumentata espressione dell’EGFR (oncogene addiction) Adeguati livelli d’inibizione dell’EGFR dose farmacocinetica tipo d’inibitore usato Alterazioni del pathway a valle dell’ EGFR (es. mutazioni attivanti K-ras, iperespressione proteine ciclo cellulare)
 Adeguati livelli d’inibizione dell’EGFR. dose. farmacocinetica. tipo d’inibitore usato. Alterazioni del pathway a valle dell’ EGFR (es. mutazioni attivanti K-ras, iperespressione proteine ciclo cellulare)")
34
X X X X X K K K K K K P P P P P P P P P Ciclina D1 Ciclina D1
Inibitore TK Inibitore TK K K K K K K P P P P P P P P P X X Recentemente è stato evidenziato un “cross-talk” tra segnali mitogenici trasdotti dal recettore dell’EGFR e ciclina D1. I segnali mitogenici trasdotti dall’EGFR inducono aumento dei livelli d’espressione della ciclina D1, così come l’inibizione funzionale dell’ EGFR si accompagna ad arresto proliferativo, accumulo delle cellule in fase G1, inibizione dell’espressione della ciclina D1 e incremento dell’espressone di p27KIP1 Larry Kalish ha ipotizzato che i livelli di ciclina D1 potessero modulare la sensibilità di cellule di HNSCC al trattamento con inibitori dell’attività TK dell’EGFR, quale il gefitinib e che l’iperespressione di ciclina D1 rendesse insensibili le cellule Ciclina D1 Ciclina D1 Ciclina D1 X proliferazione proliferazione proliferazione

35
L’iperespressione della ciclina D1 è associata a resistenza relativa nei confronti d’inibitori dell’attività TK dell’EGFR Elevati livelli d’espressione di ciclina D1 correlino con un aumentata resistenza al trattamento con gefitinib Kalish, L. H. et al. Clin Cancer Res 2004;10: Copyright ©2004 American Association for Cancer Research

36
Mutazioni dell’esone 2 del K-ras correla con la resistenza al cetuximab nei tumori del colon-retto
11% 51% Chiaramente la sensibilità all’inibizione del recettore dell’EGF dipende anche dall’integrità dei trasduttori posti a valle del recettore. Nella diapositiva si vede, infatti, come mutazioni attivanti di K-Ras siano in grado di rendere i tumori resistenti al cetuximab 5 RP + 22 SD Khambata-Ford, S. et al. J Clin Oncol; 25:

37
Bokemeyer C et al., JCO 2009

38
Come possiamo migliorare l’efficacia dei trattamenti a bersaglio molecolare ?
Strategie terapeutiche che contemplino l’utilizzo di diversi farmaci a bersaglio molecolare specifico Identificazione di fattori predittivi di risposta al trattamento con farmaci a bersaglio molecolare (es. anti-EGFR) Proprio perché i tumori sono caratterizzati dalla presenza di multiple alterazione molecolare, una strategia terapeutica promettente è l’impiego di più farmaci a bersaglio molecolare specifico, in modo da inibire i diversi pathway di segnale in gardo di promuovere la sopravvivenza e la proliferazione cellulare
 Proprio perché i tumori sono caratterizzati dalla presenza di multiple alterazione molecolare, una strategia terapeutica promettente è l’impiego di più farmaci a bersaglio molecolare specifico, in modo da inibire i diversi pathway di segnale in gardo di promuovere la sopravvivenza e la proliferazione cellulare.")
39
In questa diapositiva sono illustrati diversi farmaci a bersaglio molecolare, alcuni dei quali oggetto di studi clinici o entrati nella pratica clinica. Ci possiamo rendere conto dei progressi fatti valutando il numero di molecole disponibili. Si può ipotizzare un futuro in cui i tumori umani saranno caratterizzati non più in base alla loro derivazione istogenetica e al grado di differenziazione cellulare, ma verranno caratterizzati e trattati sulla base delle alterazione molecolari presenti

40
EGFR and VEGF patwhays:
Evidence for combining EGFR and VEGF inhibitors Interactions between EGFR and VEGF patwhays: EGFR and VEGF share common downstream signalling Endothelial cells express EGFR EGF stimulation VEGF Resistance to anti-EGFR → up-regulation of pro-angiogenetic factors Ciardiello, F. et al. Ann Oncol 2006 Ciardiello, F. et al. Clin Cancer Res 2000

41
farmaci inibitori di EGFR (cetuximab, erlotinib, gefitinib)
Tossicità cutanea farmaci inibitori di EGFR (cetuximab, erlotinib, gefitinib)
")
42
Tipo Descrizione Rash acneiforme
Eritema o eruzione maculare o papulare con o senza prurito, associato a desquamazione secca Xerosi Secchezza generalizzata della cute con fine desquamazione, fino alla comparsa di fissurazione della cute Paraonichia Eventi infiammatori dei tessuti che circondano l’unghia, fino alla formazione del granuloma piogenico

43
Tronco Giorno +192

44
Alluce piede destro Giorno +150

45
Correlation of rash and survival after treatment with ERBITUX
16 14 12 10 8 Survival (months) 6 4 2 In contrast, there did seem to be a correlation between the severity of cetuximab-induced acneiform rash and the response rate and survival time following cetuximab treatment (monotherapy or combination therapy). This relationship between rash and treatment outcome appears to be important. 99231 CRC 01412 CRC BOND3 CRC EORTC/NCI4 CRC Abbruzzese5 Pancreatic Kies6 SCCHN Study: No reaction Grade 2 Grade 1 Grade 3 1. Saltz L, et al. ASCO 2001, # 7; 2. Saltz L, et al. J Clin Oncol 2004;22:1201–1208; 3. Cunningham D, et al. N Engl J Med 2004;351:337–345; 4. Van Cutsem E, et al. EORTC/NCI Geneva 2004; 5. Abbruzzese, et al. ASCO # 518; 6. Kies, et al. ASCO 2002; # 925
 In contrast, there did seem to be a correlation between the severity of cetuximab-induced acneiform rash and the response rate and survival time following cetuximab treatment (monotherapy or combination therapy). This relationship between rash and treatment outcome appears to be important CRC CRC. BOND3. CRC. EORTC/NCI4. CRC. Abbruzzese5. Pancreatic. Kies6. SCCHN. Study: No reaction. Grade 2. Grade 1. Grade Saltz L, et al. ASCO 2001, # 7; 2. Saltz L, et al. J Clin Oncol 2004;22:1201–1208; 3. Cunningham D, et al. N Engl J Med 2004;351:337–345; 4. Van Cutsem E, et al. EORTC/NCI Geneva 2004; 5. Abbruzzese, et al. ASCO # 518; 6. Kies, et al. ASCO 2002; # 925.")
46
Overall Survival by Worst Grade of Skin Toxicity in the Panitumumab Patients
1.0 Grade 2-4 0.9 Grade 1 0.8 Hazard ratio=0.61a (95% CI: 0.40, 0.95) P =.0278 0.7 0.6 Survival Probability 0.5 0.4 0.3 0.2 0.1 0.0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Months from Randomization Patients at risk: Grade 2-4 152 150 138 120 99 71 58 39 29 17 13 9 6 5 1 Grade 1 57 55 47 34 24 20 12 8 5 4 3 2 a Hazard ratio for Grade 2-4 relative to Grade 1, stratified by ECOG and geographic region
 P = Survival Probability Months from Randomization. Patients at risk: Grade Grade a Hazard ratio for Grade 2-4 relative to Grade 1, stratified by ECOG and geographic region.")
47
Decorso della malattia
Materiale incluso in paraffina sezioni rappresentative IHC FISH Microdissezione Estrazione DNA EGFR, HER2 Ma, piuttosto, verranno caratterizzati e trattati in base alle alterazione molecolari presenti. Vi ringrazio per la cortese attenzione EGFR, HER2m pMAPK, pAKT, KI-67, ….. Stratificazione x n.copie Mutazioni EGFR, HER2, Ki-RAS qPCR-EGFR, polimorfismo introni EGFR Decorso della malattia Franklin WA, ASCO 2005










>")






 17 Ottobre 2009 DOPPIA ANTIAGGREGAZIONE PIASTRINICA.>")



 + IP = A + (g) + e ->")
>")