Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Perchè predire la struttura terziaria?
In cifre: 700,000+ sequenze proteiche ~ 20,000 strutture, ~ 5,000 uniche La distanza tra sequenze e strutture note si sta allargando. Metodi computazionali Veloci (minuti o ore), poco costosi (PC) Soluzioni corrette ca. nel 60% dei casi. Risoluzione più bassa, però spesso sufficiente per spiegare la funzione proteica Osservazione: La sequenza si evolve più rapidamente della struttura (Chothia & Lesk, 1986) Numero limitato di fold (< 1,000 ?) 100000 200000 300000 400000 500000 600000 Sequenze Comparative Models Strutture
, poco costosi (PC) Soluzioni corrette ca. nel 60% dei casi. Risoluzione più bassa, però spesso sufficiente per spiegare la funzione proteica. Osservazione: La sequenza si evolve più rapidamente della struttura (Chothia & Lesk, 1986) Numero limitato di fold (< 1,000 ) Sequenze. Comparative Models. Strutture.")
2
Sequenza-struttura-funzione/ predizione di strutture di proteine
diverse combinazioni dei 20 aa G P Y W I V R T A C D Varietà di strutture e funzioni F S Q L E K N M H ASSOCIARE A CIASCUNA PROTEINA DI CUI CONOSCIAMO LA SEQUENZA UNA O PIU’ SPECIFICHE FUNZIONI A LIVELLO MOLECOLARE

3
Codice strutturale ESPERIMENTO DI ANFINSEN
NELLA SEQUENZA PRIMARIA E’ SCRITTA LA STRUTTURA DI UNA PROTEINA PROTEINE CON SEQ. PRIMARIE SIMILI TENDONO AD AVERE STRUTTURE 3D SIMILI CONFRONTO TRA GENOMI → i geni essenziali (ciclo cell, sviluppo embrionale, signalling) soggetti a poca variabilità
 soggetti a poca variabilità.")
4
Alcune Applicazioni In primis: conoscere la struttura tridimensionale a risoluzione atomica della molecola per comprendere, spiegare, e a volte anche modificare ed utilizzare, la sua attività biologica. fisica24ore

5
Effettuare MUTAZIONI puntiformi e predire i loro effetti, che possono fornire indicazioni utili per il riconoscimento del sito attivo o di strutture indispensabili all'attività della molecola, dirette ad una certa funzione o nel matenimento della struttura della proteina. fisica24ore

6
Monitorare i cambiamenti strutturali indotti su peptidi o proteine da parte di MEMBRANE BIOLOGICHE, i quali sembrano essere fondamentali per il riconoscimento con il recettore o per oltrepassare la fase lipidica e raggiungere zone altrimenti inaccessibili. fisica24ore

7
Studiare le variazioni conformazionali provocate dall’interazione della proteina con uno o più LIGANDI, la quale fornisce l’attivazione (o inattivazione) necessaria per compiere la propria funzione biologica (o per impedirla). fisica24ore

8
Comprendere il processo di FOLDING delle proteine, ovvero il meccanismo di ripiegamento con cui raggiungono la confomazione biologicamente attiva.

9
Applicazioni FARMACOLOGICHE: viene fornita un’indicazione specifica, o quanto meno restrittiva, della struttura opportuna in funzione del bersaglio del farmaco. In questo campo, la costruzione di strutture calibrate permette di ridurre la ricerca ad un ristretto raggio d’azione.

10
Ipotesi termodinamica di Anfinsen (per proteine a singolo dominio)
L’informazione codificata nella sequenza amminoacidica di una proteina determina completamente la sua struttura nativa Lo stato nativo è il minimo assoluto dell’energia libera della proteina

11
Sequenze casuali non sono in grado di ripiegarsi in maniera univoca
Le proteine presenti in natura non sono sequenze casuali. Esse popolano un limitato gruppo di fold selezionate dalla natura attraverso l’evoluzione. Ciascuna ha un minimo globale distinto, ben separato dagli altri stati metastabili. Quali sono le proprietà uniche e comuni a tutti gli elementi di questo particolare insieme di strutture di tipo proteico?

12
Metodi per la predizione della struttura secondaria
e di domini/regioni disordinate

13
a elica foglietto b ripiegamento b aa C&F L pr Glu 1.51 1.44 ++ Val 1.70 1.49 Asn 1.56 1.28 Met 1.45 1.47 Ile 1.60 Gly 1.64 Ala 1.42 1.29 Tyr 1.25 Pro 1.52 1.91 Leu 1.21 1.30 Phe 1.38 1.32 + Asp 1.46 1.41 Lys 1.16 1.23 Trp 1.37 1.14 Ser 1.43 1.13 1.07 1.02 Cys 1.19 0.81 Gln 1.11 1.27 0.74 1.05 1.08 0.99 Thr 1.01 0.96 0.97 1.10 0.80 0.98 1.06 0.91 1.04 = Arg 0.93 0.76 His 1.00 1.22 0.89 0.95 0.88 0.87 0.68 0.83 0.82 0.90 - 0.77 0.75 0.66 0.70 0.92 0.60 0.41 0.69 0.72 0.59 0.67 0.55 0.64 -- 0.58 0.57 0.52 0.54 0.50 0.47 0.56 0.37 0.51 Ad.es. PREDIZIONE 2D che si basa sull’analisi statistica della composizione in residui delle strutture secondarie presenti nella PDB o sulle propensioni di certi amminoacidi a trovarsi in determinati elementi 2D. Propensione dei residui aminoacidici a formare elementi di struttura secondaria come riportato da Chou-Fasman (1978b) (C&F) e Levitt (1978) (L). La colonna “pr” classifica i residui come indifferenti (=) o stabilizzatori/destabilizzatori forti (++/--) e deboli (+/-) della struttura secondaria.
 (C&F) e Levitt (1978) (L). La colonna pr classifica i residui come indifferenti (=) o stabilizzatori/destabilizzatori forti (++/--) e deboli (+/-) della struttura secondaria.")
14
Metodi di predizione della struttura 2D e disordine delle proteine a partire dalla sequenza:
Diversi programmi per predizione 2D: Ad es. PSIPRED oppure JPRED che applica diversi metodi e fa un “consensus” dei vari metodi Disopred o Disoclust programmi per la predizione di regioni intrinsecamente disordinate a partire dalla sequenza. Mentre GeneSilico e’ un metaserver per la predizione del disordine usando il consenso di diversi metodi. Associata alla predizione della struttura 2D o del disordine intrinseco c’e’ sempre un livello di confidenza (da 0 a 9) che va osservato con cautela prima di trarre conclusioni. disordine
 che va osservato con cautela prima di trarre conclusioni. disordine.")
15
Diagramma di flusso della modellizzazione proteica
Sequenza proteica Dati sperimentali Ricerca nelle banchedati Allineamento multiplo di sequenza Assegnazione dei domini Proteina omologa nella banca dati PDB? Predizione della struttura secondaria/ Predizione disordine intrinseco Predizione del fold No E’ stato predetto un fold? Sì Sì Allineamento delle Sequenza ai fold noti (fold recognition) Modellizzazione Comparativa (Homology) No Predizione della struttura terziaria ab-inito Modello tridimensionale della proteina
 Modellizzazione. Comparativa (Homology) No. Predizione della. struttura terziaria ab-inito. Modello tridimensionale. della proteina.")
16
Modellizzazione comparativa (o homology modelling)
Permette di costruire la struttura tridimensionale di una proteina sulla base della SIMILARITÀ DI SEQUENZA con un’altra proteina di struttura NOTA che viene usata come STAMPO.

17
Utilizza strutture note (template) di uno o più membri di una famiglia strutturale-funzionale per predire la struttura (target) di un altro membro della famiglia la cui sequenza sia nota. Si basa sulle seguenti osservazioni: le proteine appartengono ad un numero limitato di famiglie strutturali proteine della stessa famiglia (omologhe) hanno strutture tridimensionali molto simili e conservate applicabile con successo se esiste almeno un omologa a struttura nota (id. > 30% con il templato)
 hanno strutture tridimensionali molto simili e conservate. applicabile con successo se esiste almeno un omologa a struttura nota (id. > 30% con il templato)")
18
HOMOLOGY MODELLING OMOLOGO 3D (PDB) ALLINEAMENTO RICERCA DEL TEMPLATO
Blast-FastA CRITERI IDENTITA’/SIMILARITA’ CONOSCENZA FUNZ.-STR.-BIOCHIM.

19
ALLINEAMENTO GUIDA LA COSTRUZIONE DEL MODELLO
CORRISPONDENZA aa target aa templato ricerca ALLINEAMENTO OTTIMALE CORRISPONDENZA DI aa FUNZ. IMPORTANTI CORRISPONDENZA DELLA STRUTTURA SECONDARIA TRA TEMPLATO E QUERY VALUTAZIONE DEI GAP loop USO TEMPLATI MULTIPLI loc.similarità

20
ALLINEAMENTO 1. Generazione di allineamenti a coppie diversi con diversi programmi (nel caso di un solo templato) 2. Allineamenti multipli di omologhi per avere informazioni maggiori 3. Ricerca di informazioni biologico-biochimiche sugli aa conservati 4. Predizione di struttura secondaria per il target 5. Correzione dell’allineamento sulla base delle informazioni ai punti e 4.

21
Dalla sequenza al modello
Raw model Loop modeling Side chain placement Refinement

22
CREAZIONE DEL MODELLO ______________ ______________ x-ray SCRs
identificazione SCR (structural conserved regions) SCR scaffold del modello Raw model Loop modeling Side chain placement Refinement ______________ ______________ x-ray SCRs No SCRs (loops ?)
 SCR scaffold del modello. Raw model. Loop modeling. Side chain placement. Refinement. ______________. ______________ x-ray. SCRs. No SCRs (loops )")
23
Costruzione del pre-modello
La struttura del templato viene utilizzata come “stampo“ per costruire il modello seguendo l‘allineamento. Le coordinate 3D dei residui strutturalmente conservati si possono copiare direttamente. Le regioni variabili della struttura (generalmente loop) non si possono copiare. flexible conserved
 non si possono copiare. flexible. conserved.")
24
Catene laterali Problema: Applicando le coordinate del templato sulla sequenza del target cambiano tipo, dimensione e posizione delle catene laterali. L‘RMSD cambia relativamente poco, però possono cambiare le conformazioni di residui importanti (p.es. del sito attivo) Dove possibile è meglio mantenere le conformazioni delle catene laterali del templato. Esistono metodi standard per risolvere questo problema. Raw model Loop modeling Side chain placement Refinement
 Dove possibile è meglio mantenere le conformazioni delle catene laterali del templato. Esistono metodi standard per risolvere questo problema. Raw model. Loop modeling. Side chain placement. Refinement.")
25
PREDIZIONE DELLE CATENE LATERALI
AUSILIO DI LIBRERIE DI ROTAMERI Contengono i possibili conformeri delle catene laterali a fronte di specifiche conformazioni del backbone OTTIMIZZAZIONE ENERGETICA DELLE STRUTTURA rimozione di clash

26
Loop modeling Al pre-modello possono mancare interi frammenti di catena principale non conservati nella famiglia proteica Inserzioni Delezioni Descrizione del problema: Si cerca un fold che colleghi il frammento N-terminale (pre-loop) con quello C-terminale (post-loop) tramite k residui (f,y) sono gli unici parametri liberi Raw model Loop modeling Side chain placement Refinement loop post-loop pre-loop
 con quello C-terminale (post-loop) tramite k residui. (f,y) sono gli unici parametri liberi. Raw model. Loop modeling. Side chain placement. Refinement. loop. post-loop. pre-loop.")
27
PREDIZIONE DEI LOOP REGIONI VARIABILI – pressione selettiva
DUE STRATEGIE PREDITTIVE: criterio geometrico testando diverse conformazioni ricerca in PDB di frammenti simili nelle proteine a struttura nota INFLUENZA DELL’INTORNO REFINEMENT CRITICO OTTIMIZZAZIONE MEC. E DIN. MOL. CRITICITA’ nella predizione quando i LOOP rivestono ruolo funzionale o di interazione.

28
5. Ottimizzazione del modello
Regolarizzazione di legami, angoli e torsioni Eliminazioni di clash strutturali Minimizzazione energetica

29
6. Controllo della qualità del modello
programmi che valutano la qualità dal punto di vista geometrico della struttura (grafico di ramachandran, planarità, clash…) programmi che fanno una predizione dell’rmsd del modello della proteina rispetto alla sua putativa struttura nativa
 programmi che fanno una predizione dell’rmsd del modello della proteina rispetto alla sua putativa struttura nativa.")
30
FOLD RECOGNITION Predizione di sequenza con similarità con proteine a struttura nota. Osservazione: La natura utilizza solamente un numero limitato di fold diversi Idea della fold recognition: Cerca di rappresentare la struttura ignota con dei fold conosciuti, valuta quale potrebbe essere quello “corretto“.

31
FOLD RECOGNITION per casi predittivi in cui non ci sono omologie chiare con proteine a struttura nota (TWILIGHT ZONE (id.seq tra target e templato 15<x<30%) metodi che rinunciano alla corretta formulazione del campo di forze agenti su una struttura proteica detti mean force potential che individuano un potenziale che cattura la natura risultante delle forze in gioco devo disporre di uno strumento quantitativo per misurare fitness di una sequenza con una struttura per poter assegnare alla sequenza in questione le strutture note e valutare la bontà dell’assegnazione queste funzioni di pseudo-potenziale sono costruite sulla base di un’analisi statistica di strutture note e sono la base che differenzia i diversi metodi di fold-recognition
 metodi che rinunciano alla corretta formulazione del campo di forze agenti su una struttura proteica. detti mean force potential che individuano un potenziale che cattura la natura risultante delle forze in gioco. devo disporre di uno strumento quantitativo per misurare fitness di una sequenza con una struttura per poter assegnare alla sequenza in questione le strutture note e valutare la bontà dell’assegnazione. queste funzioni di pseudo-potenziale sono costruite sulla base di un’analisi statistica di strutture note e sono la base che differenzia i diversi metodi di fold-recognition.")
32
METODO DEGLI “Structural environment”
detto anche metodo dei profili 1D-3D o di Eisenberg ASSUNZIONE: ambiente di un residuo più conservato del residuo stesso (adatto a relazioni distanti) descrive intorno strutturale di ogni aa in ogni proteina a struttura nota e valuta la frequenza con cui ognuno dei 20 aa si trova in un certo intorno ambiente/intorno strutturale è descritto sulla base di 3 principi (aspetti + importanti di una struttura proteica): Quanto un residuo in una posizione è schermato dal solvente? Quanto è a contatto con gruppi polari o idrofobici? La struttura secondaria in cui è collocato Combinazioni dei 3 parametri definiscono diverse CATEGORIE STRUTTURALI (in tutto 18 classi strutturali) 6 classi rappresentano l’intorno delle catene laterali che può essere non accessibile (B), parzialmente accessibile (P) o accessibile al solvente (E) P e B sono suddivisi sulla base della frazione dell’intorno che è costituita da atomi polari 1– 3 categorie di frazioni di contatti polari (frazione dell’area della catena laterale coperta da atomi polari): 0-0.4, , > E sempre a elevata frazione polare. 6 classi: E, P1, P2, B1, B2 e B3 Ciascuna di queste potrà trovarsi in una struttura α o β o “altro” e quindi le classi possibili sono 18.
 descrive intorno strutturale di ogni aa in ogni proteina a struttura nota e valuta la frequenza con cui ognuno dei 20 aa si trova in un certo intorno. ambiente/intorno strutturale è descritto sulla base di 3 principi (aspetti + importanti di una struttura proteica): Quanto un residuo in una posizione è schermato dal solvente Quanto è a contatto con gruppi polari o idrofobici La struttura secondaria in cui è collocato. Combinazioni dei 3 parametri definiscono diverse CATEGORIE STRUTTURALI (in tutto 18 classi strutturali) 6 classi rappresentano l’intorno delle catene laterali che può essere non accessibile (B), parzialmente accessibile (P) o accessibile al solvente (E) P e B sono suddivisi sulla base della frazione dell’intorno che è costituita da atomi polari 1– 3 categorie di frazioni di contatti polari (frazione dell’area della catena laterale coperta da atomi polari): 0-0.4, , > 0.8. E sempre a elevata frazione polare. 6 classi: E, P1, P2, B1, B2 e B3. Ciascuna di queste potrà trovarsi in una struttura. α o β o altro e quindi le classi possibili sono 18.")
33
METODO DEGLI “Structural environment”
A questo punto entra in gioco un eventuale sequenza x cui voglio valutare la fitness per una data struttura (sequenza target). Confronterò gli amminoacidi della mia sequenza con gli ambienti strutturali definiti sulla base delle strutture note (si parla in questo senso di ALLINEAMENTO SEQUENZA-STRUTTURA) Si cerca, in sostanza, di adattare ad un fold già noto la sequenza in esame e si cerca l’allineamento ottimale (operazione fatta tra la seq e tutte le strutture disponibili); All’allineamento è sempre assegnato un punteggio di cui valutare la significatività statistica (E-value o z-score) .
. Confronterò gli amminoacidi della mia sequenza con gli ambienti strutturali definiti sulla base delle strutture note (si parla in questo senso di ALLINEAMENTO SEQUENZA-STRUTTURA) Si cerca, in sostanza, di adattare ad un fold già noto la sequenza in esame e si cerca l’allineamento ottimale (operazione fatta tra la seq e tutte le strutture disponibili); All’allineamento è sempre assegnato un punteggio di cui valutare la significatività statistica (E-value o z-score) .")
34
METODO DEI CONTATTI TRA AMMINOACIDI
Si basa, per descrivere i fold noti, su interazioni tra coppie di amminoacidi (sulla distanza tra amminoacidi nelle proteine a struttura nota); ASSUNZIONE ALLA BASE: le strutture native delle proteine sono stabilizzate da interazioni intramolecolari tra i vari atomi della proteina e intermolecolari tra essi e le molecole di solvente Dalla banca dati di strutture costruisce una funzione pseudopotenziale che descrive lo spazio conformazionale della proteina: per ogni possibile coppia di aa in una struttura valuto la distanza (di solito Cα-Cα) tra i residui in esame e calcolo una funzione che descrive questa interazione; Le funzioni pseudopotenziale sono una per ogni coppia di residui e le costruisco sulla base della frequenza con cui osservo una data coppia a una data distanza nel database PDB.
; ASSUNZIONE ALLA BASE: le strutture native delle proteine sono stabilizzate da interazioni intramolecolari tra i vari atomi della proteina e intermolecolari tra essi e le molecole di solvente. Dalla banca dati di strutture costruisce una funzione pseudopotenziale che descrive lo spazio conformazionale della proteina: per ogni possibile coppia di aa in una struttura valuto la distanza (di solito Cα-Cα) tra i residui in esame e calcolo una funzione che descrive questa interazione; Le funzioni pseudopotenziale sono una per ogni coppia di residui e le costruisco sulla base della frequenza con cui osservo una data coppia a una data distanza nel database PDB.")
35
METODO DEI CONTATTI TRA AMMINOACIDI
Ad esempio x la coppia ala-ala: misuro in ogni proteina la distanza tra i Cα e costruisco una distribuzione in cui metto in relazione ogni distanza alla frequenza con cui è osservata Per passare dalle frequenze a energie si usa il principio di Boltzmann che correla probabilità di uno stato con la sua energia La distanza geometrica r è influenzata dalla distanza in sequenza: si prende in considerazione anche questa; Parametri da considerare: a,b (i 2 residui); c,d (atomi considerati per le distanze, es Cα-Cα); k è la distanza in sequenza.
; c,d (atomi considerati per le distanze, es Cα-Cα); k è la distanza in sequenza.")
36
Threading/Fold-recognition
Homology modelling Threading/Fold-recognition Identifica prima gli omologhi Prova tutte le possibili strutture Si determina l’allineamento ottimale Prova tutti i possibili allineamenti strutturali Ottimizza un modello Valuta molti modelli poco accurati nei dettagli per F.RECOGNITION/THREADING: Phyre, GenThreader, 3D-PSSM + metaserver che fanno consenso per HOMOLOGY MODELLING: programma Modeller HHPRED server: sia per homology modelling che threading in caso di templati con identita’ non troppo bassa (perche’ si appoggia per definire gli allineamenti su metodi accurati per definire omologie remote tipo HMM

37
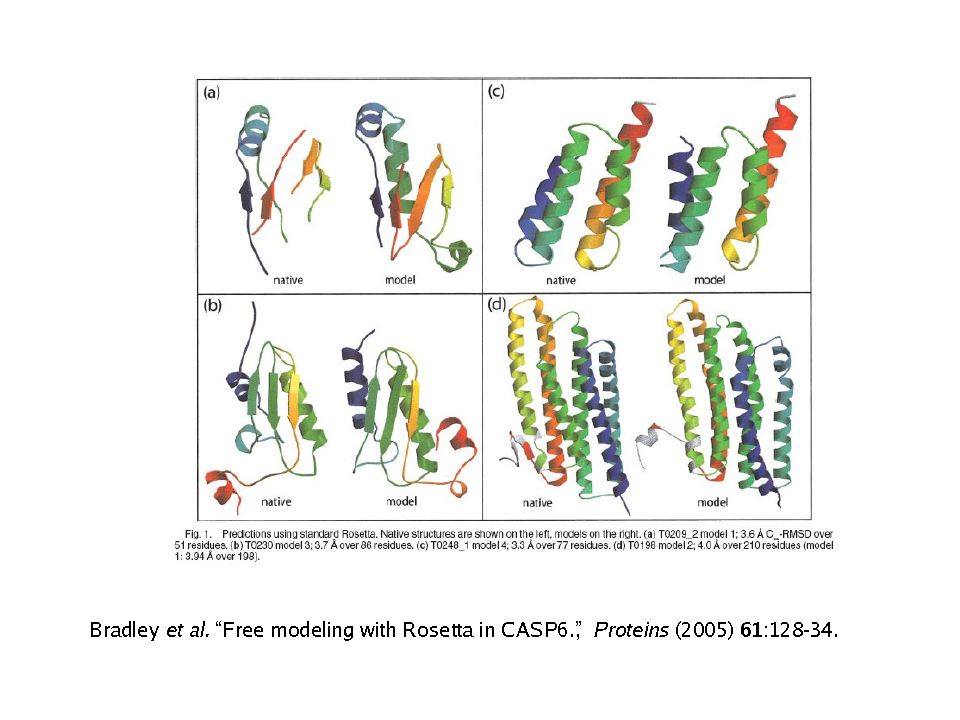
Il metodo ab initio (predizione de novo)
Il problema della predizione di struttura ab initio “data una sequenza proteica, calcolarne la struttura” Il calcolo è basato sulla stima dell’energia relativa alla posizione di ciascun atomo nello spazio e la sua relazione chimico-fisica con gli altri atomi Il minimo globale della funzione energia definisce la struttura 3D È teoricamente possibile Essendo la biofisica complessa ed incompleta è nella pratica ancora molto difficile ROSETTA, sviluppata dal gruppo di David Baker

38
Ab initio methods for modelling
NO allineamento NO struttura nota Costruire una funzione empirica che descriva le forze di interazione Esplorare lo spazio conformazionale per massimizzare funzione di merito Programmi per ab-initio es. Rosetta o I-Tasser (I-Tasser usa metodi misti di threading e ab-initio) 31
 31.")
39
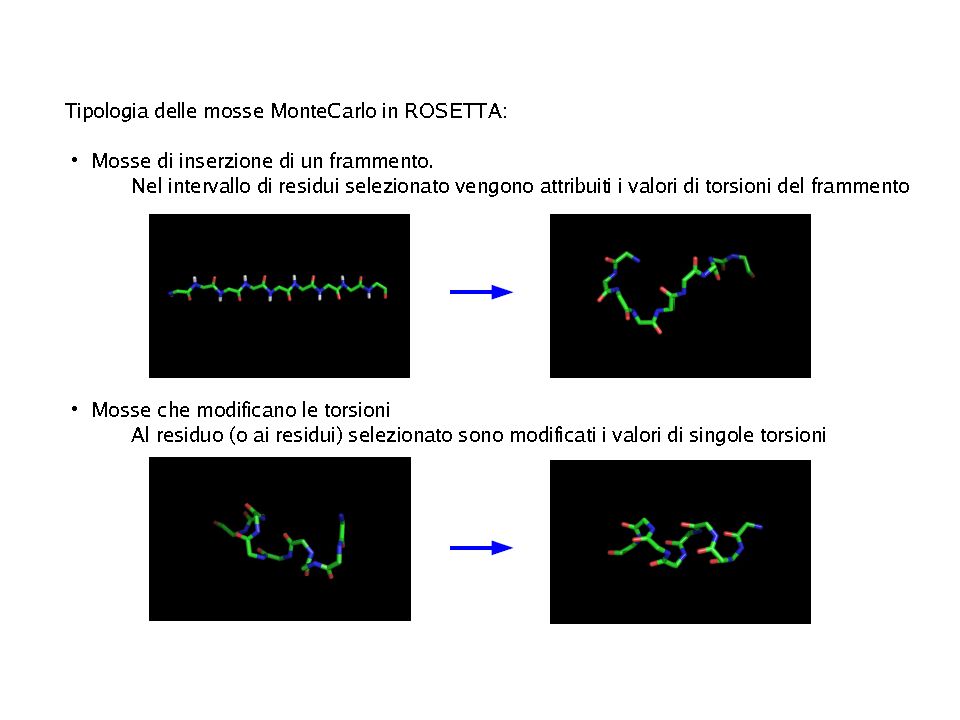
“Modello di folding” su cui si basa Rosetta:
“ Local sequence fragments rapidly alternate between different possible local structures, and folding occurs when the conformations and relative orientations of these local segments combine to form low energy global structures” E’ UN PROBLEMA DI CAMPIONAMENTO CONFORMAZIONALE

40
PROBLEMA DI CAMPIONAMENTO CONFORMAZIONALE:
Ricerca della geometria di minima energia Richiede: Metodo di ricerca/generazione delle conformazioni Funzione “energetica” (funzione di scoring energetico) per valutarle Nei metodi ab-initio: Rappresentazioni ridotte delle proteine/ Potenziali detti knowledge-based semplificati/ ricerca di conformazionale coarse-grain
 per valutarle. Nei metodi ab-initio: Rappresentazioni ridotte delle proteine/ Potenziali detti knowledge-based semplificati/ ricerca di conformazionale coarse-grain.")
46
Building by homology (Homology modelling)
Allineamento con proteine a struttura nota - G Y M A K S T F L E D V I Modello strutturale 4

47
S L V A Y G M Fold recognition (Threading) Sequenza: +
Motivi strutturali noti S L V A Y G M Modello strutturale 5

48
Ab initio Sequenza S L V A Y G M Modello strutturale 6

49
NOTE GENERALI PER IL MODELLING
prima di cercare di ottenere un modello raccogliere quante piu’ informazioni possibili sulla proteina, e soprattutto definire i suoi domini strutturali (che potrebbero richiedere procedure di modelling diverse) raccogliere informazioni su propensione a determinate strutture secondarie e disordine intrinseco valutare con metodi di ricerca di similarita’ in banche dati (Blast, PSI-Blast) se esistono omologhe a struttura nota Fare allineamenti multipli con le omologhe a struttura nota e eventualmente altre a sola sequenza nota per identificare bene regioni conservate nella famiglia di proteine di studio Validare sempre gli allineamenti proposti dai programmi per il modelling (soprattutto per metodi di threading e ab-initio o per casi di templati con bassa identita’ di sequenza)
 raccogliere informazioni su propensione a determinate strutture secondarie e disordine intrinseco. valutare con metodi di ricerca di similarita’ in banche dati (Blast, PSI-Blast) se esistono omologhe a struttura nota. Fare allineamenti multipli con le omologhe a struttura nota e eventualmente altre a sola sequenza nota per identificare bene regioni conservate nella famiglia di proteine di studio. Validare sempre gli allineamenti proposti dai programmi per il modelling (soprattutto per metodi di threading e ab-initio o per casi di templati con bassa identita’ di sequenza)")
Presentazioni simili








![PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]](/1/540371/big_thumb.jpg "PROSITE contiene anche pattern ad ALTA OCCORRENZA, corti e aspecifici (modifiche post-traduzionali) Es. phosphorylation by CK2 [ST]-x(2)-[DE]>")













