Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
GENITALI AMBIGUI SINDROME ADRENOGENITALE
Manuela Caruso Dipartimento di Pediatria, Università di Catania

2
GENITALI AMBIGUI Si definiscono ambigui i genitali esterni con caratteristiche tali che non permettono una sicura attribuzione di sesso

3
GENITALI AMBIGUI Ipertrofia clitoridea - grandi labbra nella norma
- parziale fusione grandi labbra - seno urogenitale Formazione similpeniena - scroto disabitato Ipospadia Micropene Micropene e scroto disabitato Agenesia del pene Criptorchidismo

4
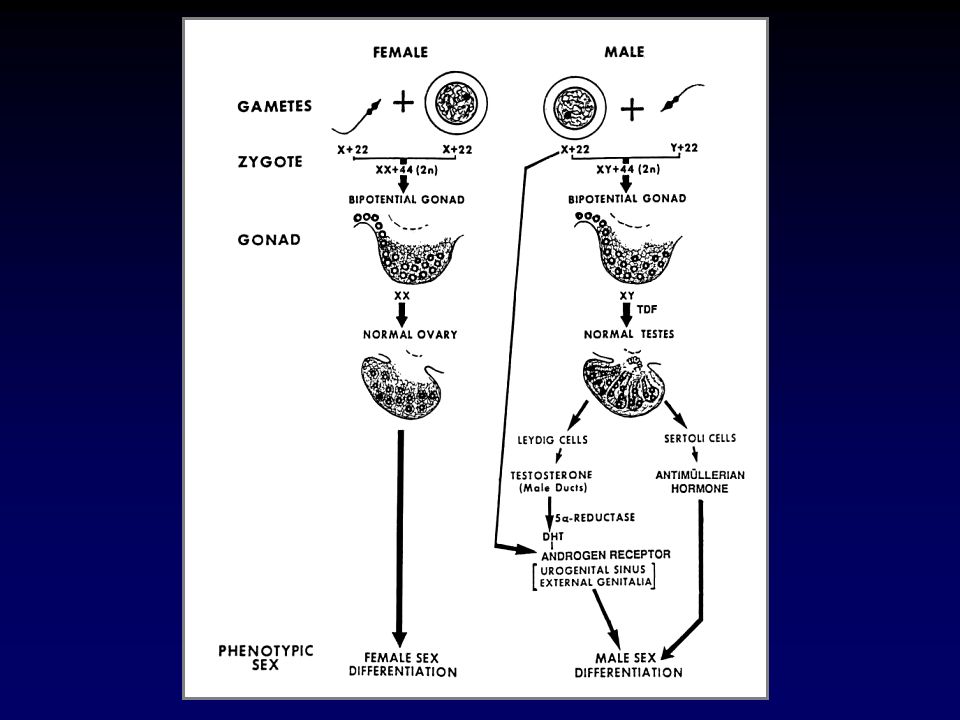
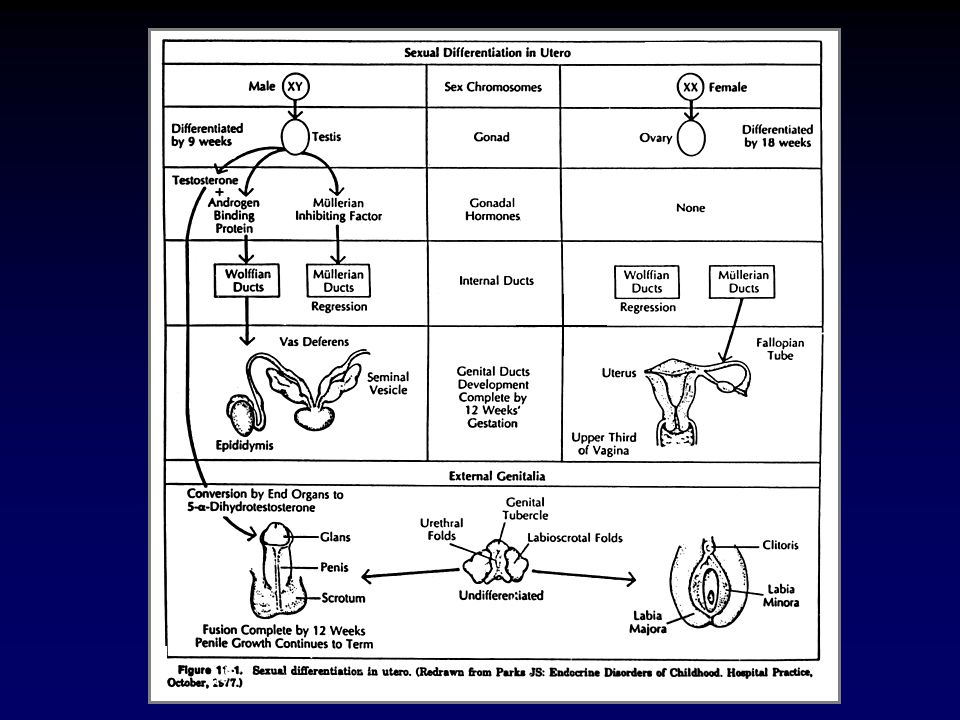
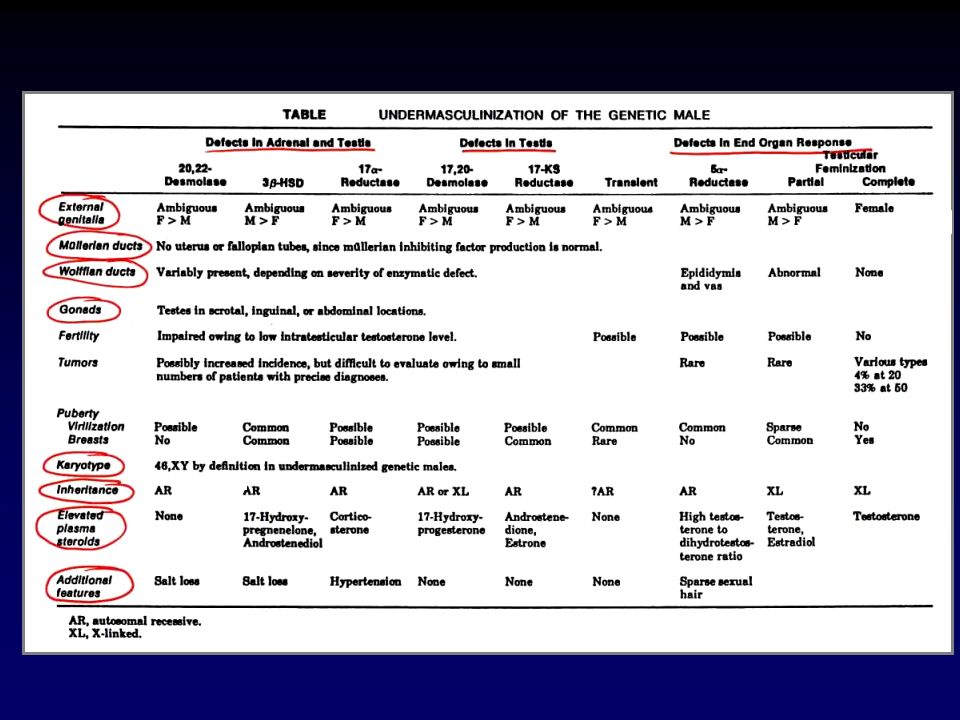
CAUSE DI GENITALI AMBIGUI
Virilizzazione di una femmina XX Incompleta mascolinizzazione di un maschio XY Anomalie gonadiche e/o cromosomiche Difetti embriogenetici non attribuibili ad anomalie gonadiche o ormonali

12
Trasposizione penoscrotale Pene bifido con estrofia vescicale
DIFETTI EMBRIOGENETICI NON ATTRIBUIBILI AD ANOMALIE GONADICHE O ORMONALI Epispadia Trasposizione penoscrotale Pene bifido con estrofia vescicale Agenesia peniena associata ad ano imperforato

13
APPROCCIO DIAGNOSTICO
Anamnesi Patologia materna/assunzione farmaci Familiarità per genitali ambigui Familiarità per bassa statura Familiarità per irsutismo Prelievo per: Cariotipo alla nascita Elettroliti Attività reninica Testosterone DHT Ritmo del cortisolo 17-OH-progesterone dopo il 5°giorno di vita DHEAS 17βestradiolo LH – FSH Controllo pressione arteriosa

14
FEMMINA 46, XX Cortisolo Renina Nella norma Causa materna No Si DHEAS
Nα K T 17- OHP Renina Nα K 17 - OHP CAH OH CAH β - OH Ermafro-ditismo Biopsia gonadi

15
INQUADRAMENTO DIAGNOSTICO MASCHIO
46, XY Cortisolo T Nα K CAH β - OL LH Ipoplasia cellule di Leydig CAH OH DHEAS Renina PA Nella norma Biopsia gonadica DHT Ermafrodismo o disgesia gonadica Deficit 5α Reduttasi

17
SINDROME ADRENOGENITALE (SAG) IPERPLASIA SURRENALE CONGENITA
La SAG è dovuta a deficit enzimatici che determinano una ridotta efficienza nella sintesi del cortisolo L’aumentata stimolazione del surrene ad opera dell’ACTH, al fine di aumentare i livelli di cortisolemia, determina un’iperplasia della ghiandola e un eccesso dei metaboliti a monte del blocco enzimatico L’attività biologica dei precursori e dei loro metaboliti prodotti in eccesso e il deficit degli ormoni a valle del blocco enzimatico, determineranno il quadro clinico della SAG

18
3β-HSD 11β-OH 18-OH deidrogenasi Androstenedione Deidroepiandrosterone

19
DIFETTI ENZIMATICI RESPONSABILI DI SAG
Deficit di 21 - idrossilasi → % Deficit di 11β - idrossilasi Deficit di 17α - idrossilasi Deficit di 3β - ol deidrogenasi Prevalenza SAG : 1: – 1:20.000

20
Manifestazioni cliniche nei vari tipi di iperplasia surrenalica congenita
* Genitali femminili normali nella forma ad esordio tardivo e nella forma criptica ** Clitoridomegalia

21
FORME CLINICHE DELLA SAG DA DEFICIT DI 21-OH
SAG classica virilizzante semplice 25% SAG classica con perdita di Sali 75% SAG non classica (SAGNC) a insorgenza tardiva (SAGIT) “late onset”
 a insorgenza tardiva (SAGIT) late onset")
22
3β-HSD 3β-HSD 3β-HSD 11β-OH 11β-OH 18-OH deidrogenasi
Deidroepiandrosterone 3β-HSD 3β-HSD 3β-HSD Androstenedione 11β-OH 11β-OH 18-OH deidrogenasi

23
SAG DA DEFICIT DI 21-OH Forma Virilizzante Semplice ♀
Nella femmina l’esposizione intrauterina ad elevati livelli di androgeni provoca la virilizzazione dei genitali esterni La neonata si presenta con un grado di virilizzazione variabile dall’ipertrofia clitoridea ad un quadro di pseudoermafroditismo femminile (Stadi di Prader 1→5) e con genitali interni del tutto normali La SAG da deficit di 21-OH è la causa più frequente di ambiguità genitale La paziente non trattata presenterà un progressivo aggravamento della virilizzazione e difficoltà (shock) ad affrontare le condizioni di stress
 e con genitali interni del tutto normali. La SAG da deficit di 21-OH è la causa più frequente di ambiguità genitale. La paziente non trattata presenterà un progressivo aggravamento della virilizzazione e difficoltà (shock) ad affrontare le condizioni di stress.")
24
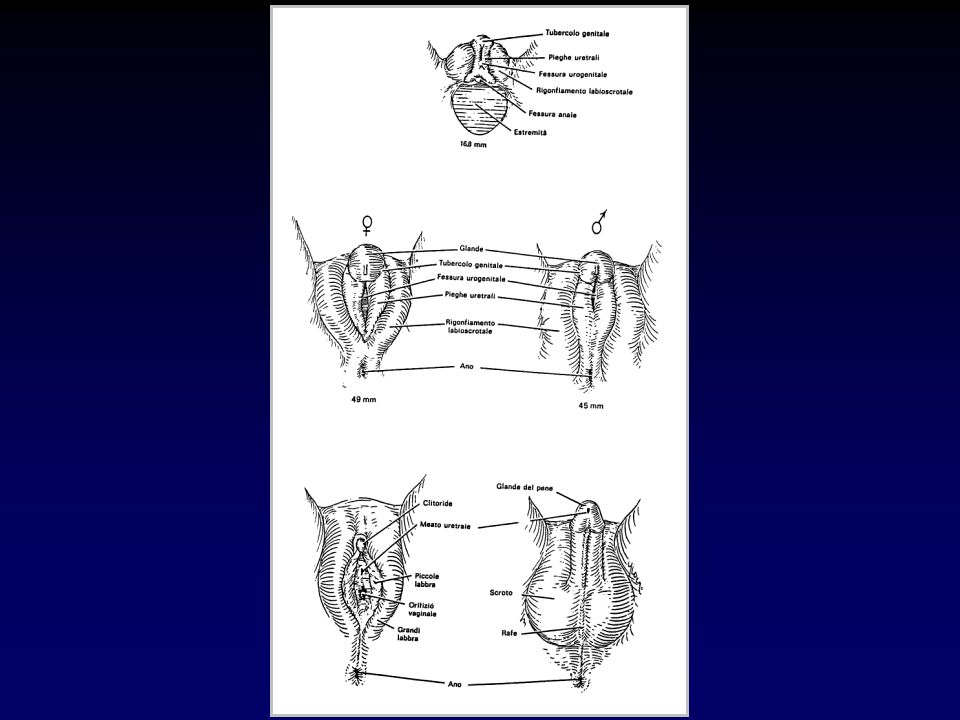
STADI DI INTERSESSUALITÀ SECONDO PRADER
- Stadio I: ipertrofia clitoridea - Stadio II: gradi variabili di fusione delle grandi labbra - Stadio III: orifizio vaginale in comune con l’uretra (seno urogenitale) - Stadi IV e V: uretra fallica con meato esterno penoscrotale o all’estremità del pene e fusione completa delle grandi labbra
 - Stadi IV e V: uretra fallica con meato esterno penoscrotale o all’estremità del pene e fusione completa delle grandi labbra.")
25
AMBIGUITÀ DEI GENITALI

26
SAG DA DEFICIT DI 21-OH Forma Virilizzante Semplice ♂
Il neonato maschio presenta genitali esterni normali, può essere riscontrata una macrogenitosomia L’aumentata esposizione agli androgeni si manifesterà successivamente (3-5 anni) con il quadro clinico della pseudopubertà precoce La SAG da deficit di 21-OH è la causa più frequente di pseudopubertà precoce Inoltre, il paziente sarà a rischio di shock in condizioni di stress (ipocortisolemia)
 con il quadro clinico della pseudopubertà precoce. La SAG da deficit di 21-OH è la causa più frequente di pseudopubertà precoce. Inoltre, il paziente sarà a rischio di shock in condizioni di stress (ipocortisolemia)")
27
PSEUDOPUBERTÁ PRECOCE ♂
Accelerazione della crescita Accelerazione della maturazione ossea Peluria pubica Ingrossamento del pene Aumento della pigmentazione e della rugosità della borsa scrotale Testicoli di dimensioni prepuberali Acne Ipertrofia muscolare Modificazione del timbro della voce

28
SAG DA DEFICIT DI 21-OH Forma Virilizzante Semplice ♂
Pseudopubertà precoce

29
SAG DA DEFICIT DI 21-OH Forma con perdita di sali
E’ un’emergenza neonatale con elevato rischio per la vita Il quadro clinico è dovuto ad un grave squilibrio idroelettrolitico E’ dovuta a carenza di attività mineralcorticoide

30
3β-HSD 3β-HSD 3β-HSD 11β-OH 11β-OH 18-OH deidrogenasi
Deidroepiandrosterone 3β-HSD 3β-HSD 3β-HSD Androstenedione 11β-OH 11β-OH 18-OH deidrogenasi

31
SAG DA DEFICIT DI 21-OH Forma con perdita di sali
Il quadro clinico si manifesta tra la 2° e la 3° settimana di vita ed è caratterizzato da: Scarso appetito Arresto dell’accrescimento/calo ponderale Vomito Segni di disidratazione Letargia

32
SAG DA DEFICIT DI 21-OH Forma con perdita di sali
In pochi giorni il quadro clinico evolverà con comparsa di: Disturbi del ritmo Dispnea Cianosi Acidosi Ipotensione Collasso → Decesso

33
SAG DA DEFICIT DI 21-OH Forma con perdita di sali
ATTENZIONE!! Nella femmina la virilizzazione più o meno marcata avrà già permesso di formulare la diagnosi o almeno il sospetto diagnostico Nel maschio essendo normali i genitali, la perdita di Sali rappresenta la prima manifestazione della malattia RISCHIO!! La diagnosi differenziale include: sepsi, stenosi del piloro, gastroenterite, ipoplasia congenita del surrene, pseudoipoaldosteronismo, cardiopatie congenite

34
SAG DA DEFICIT DI 21-OH SOSPETTO DIAGNOSTICO Genitali ambigui
Macrogenitosomia Sintomi e segni di perdita di sali ESAMI DIAGNOSTICI 17-OH progesterone (↑), testosterone (↑) Na (↓), K (↑), PRA (↑) CONFERMA DIAGNOSI Studio molecolare gene CYP21 Mutazioni responsabili circa 50
, testosterone (↑) Na (↓), K (↑), PRA (↑) CONFERMA DIAGNOSI. Studio molecolare gene CYP21. Mutazioni responsabili circa 50.")
35
TERAPIA DELLA SAG DA DEFICIT DI 21-OH
TERAPIA DI ATTACCO (PERDITA DI SALI) Idrocortisone e.v in infusione mg/m2/die Desossicortone i.m. 1 mg/kg/die Terapia infusionale (glucosata,salina) TERAPIA DI MANTENIMENTO Idrocortisone p.o mg/m2 in 3 somministrazioni o Cortisone acetato p.o mg/m2 Fludrocortisone p.o mg/die in 1 somministrazione Nei primi 2 anni di vita supplementare con 2 gr di NaCl/die
 Idrocortisone e.v in infusione mg/m2/die. Desossicortone i.m. 1 mg/kg/die. Terapia infusionale (glucosata,salina) TERAPIA DI MANTENIMENTO. Idrocortisone p.o mg/m2 in 3 somministrazioni o. Cortisone acetato p.o mg/m2. Fludrocortisone p.o mg/die in 1 somministrazione. Nei primi 2 anni di vita supplementare con 2 gr di NaCl/die.")
36
TERAPIA DELLA SAG DA DEFICIT DI 21-OH
TERAPIA IN CASO DI STRESS In caso di stress, febbre, interventi chirurgici, infezioni, affezioni gastrointestinali acute la dose di cortisone va raddoppiata o triplicata In caso di vomito ripetuto la terapia va somministrata per via i.m. TERAPIA NELL’ADOLESCENTE In caso di difficoltà a sopprimere l’asse con la terapia classica o di cattiva compliance si può usare il desametazone a 0.25 mg/m2 /die in singola somministrazione

37
SAG NON CLASSICA INSORGENZA TARDIVA / LATE ONSET
Quadri di iperandrogenismo ad insorgenza postnatale Sintomatologia estremamente variabile che può comparire ad ogni età E’ dovuta a mutazioni lievi con parziale riduzione dell’attività enzimatica

38
SAG NON CLASSICA INSORGENZA TARDIVA / LATE ONSET
Pubarca prematuro Accelerazione velocità di crescita e maturazione ossea Clitoridomegalia Acne Irsutismo Oligomenorrea Alopecia PCOS Infertilità

39
Spettro delle manifestazioni cliniche delle SAG da deficit di 21-OH

40
AUTOSOMAL RECESSIVE GENETIC DISORDES NONCLASSICAL 21-HYDROXYLASE
DISEASE FREQUENCY: AUTOSOMAL RECESSIVE GENETIC DISORDES 0.2 0.1 0.01 0.001 0.0001 X Ashkenazi Jews Yugoslavs Hispanics Italians Sum of Ethnic groups Yupik Eskimos Caucasians (Italians) Mixed Non-Jewish Caucasian NONCLASSICAL 21-HYDROXYLASE DEFICIENCY CLASSICAL 21-HYDROXYLASE Phenylketonuria X Tay-Sachs (Ashkenazi Jews) Cystic Fibrosis Sickle cell Anemia (Blacks) Speiser PW et al Am J Hum Genet 1985
 Mixed. Non-Jewish. Caucasian. NONCLASSICAL 21-HYDROXYLASE. DEFICIENCY. CLASSICAL. 21-HYDROXYLASE. Phenylketonuria. X Tay-Sachs. (Ashkenazi Jews) Cystic. Fibrosis. Sickle cell. Anemia. (Blacks) Speiser PW et al Am J Hum Genet")
Presentazioni simili





>")


















