Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

2
Bisogni e problemi delle cure primarie
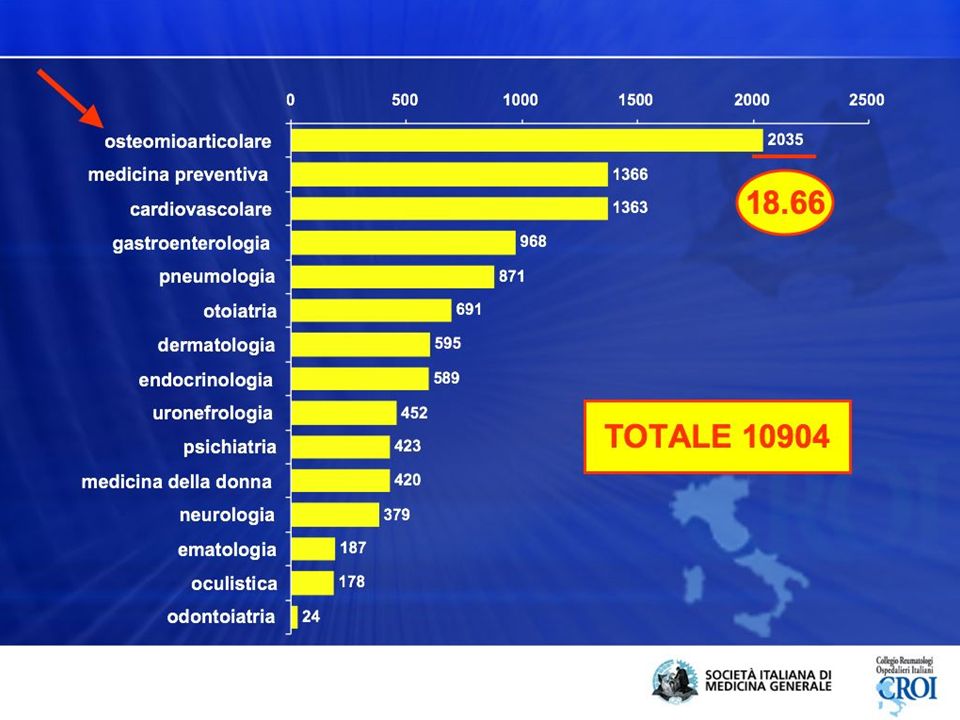
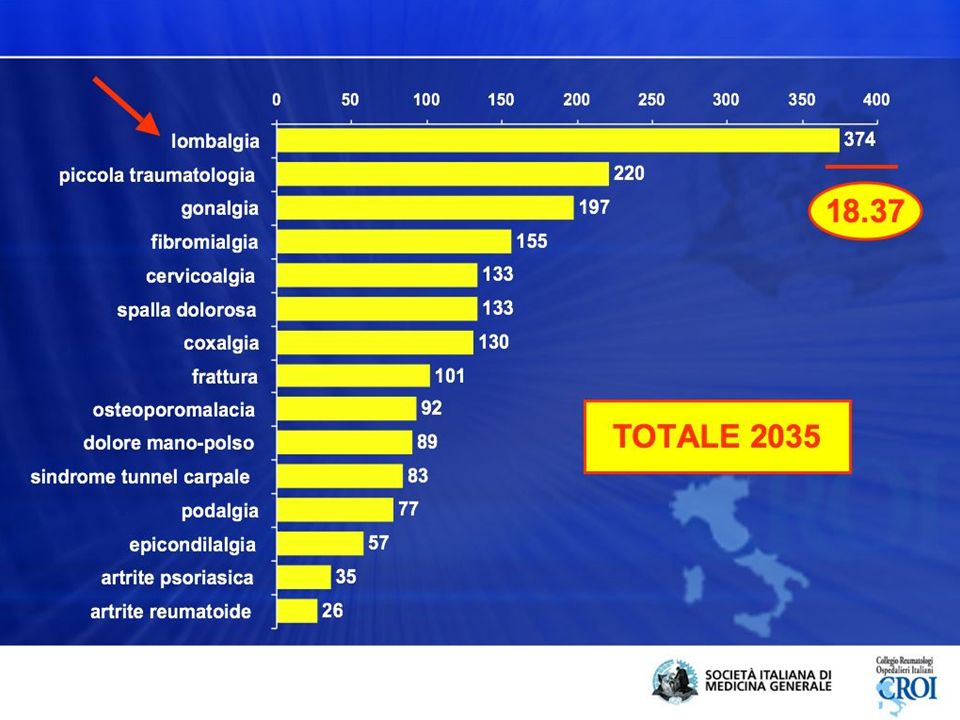
inquadramento Bisogni e problemi delle cure primarie 1 anno di lavoro di un MMG con 1500 pazienti 10944 accessi totali al servizio MG 5331 presso il personale di segreteria/infermiera 5613 visite mediche 10904 problemi affrontati

5
2. CONNETTIVITI E VASCULITI (MALATTIE REUMATICHE SISTEMICHE) 2.1
Lupus eritematoso Lupus eritematoso sistemico Lupus associato a deficit congeniti del complemento Lupus indotto da farmaci * Lupus discoide fisso Lupus cutaneo subacuto Lupus neonatale 2.2. Sindromi sclerodermiche Sclerosi sistemica Sclerodermia circoscritta Morfea Sclerodermia lineare Fascite diffusa con o senza eosinofilia Sindromi sclerodermiche da agenti ambientali e chimici Sindrome eosinofilia – mialgia Scleredema Scleromixedema 2.3 Miositi Polimiosite / dermatomiosite Dermatomiosite (Polimiosite) associata a neoplasia Miosite da corpi inclusi Dermatomiosite amiopatica Altre forme di miosite Miosite granulomatosa Miosite eosinofilica Miosite focale o nodulare Miosite ossificante 2.4. Sindrome di Sjögren e forme correlate Sindrome di Sjögren primitiva Sindrome di Sjögren associata ad altre malattie autoimmuni Sindrome sicca associata ad altre malattie
 associata a neoplasia. Miosite da corpi inclusi. Dermatomiosite amiopatica. Altre forme di miosite. Miosite granulomatosa. Miosite eosinofilica. Miosite focale o nodulare. Miosite ossificante Sindrome di Sjögren e forme correlate. Sindrome di Sjögren primitiva. Sindrome di Sjögren associata ad altre malattie autoimmuni Sindrome sicca associata ad altre malattie")
6
2.5. Sindromi da sovrapposizione (overlap) Connettivite mista
Connettivite mista Polimiosite – Sclerodermia Lupus eritematoso sistemico – Sindrome di Sjögren * Sindrome Rhupus (Artrite reumatoide – Lupus eritematoso sistemico) Sclerolupus (Sclerosi sistemica – Lupus eritematoso sistemico) Sclerosi sistemica – Cirrosi biliare primitiva – Sindrome di Sjögren Altre sindromi da sovrapposizione 2.6. Connettiviti indifferenziate 2.7. Vasculiti sistemiche (Primitive) Arterie di grosso e medio calibro Arterite di Takayasu Arterite gigantocellulare di Horton Arterie di medio calibro Panarterite nodosa Vasi di medio e piccolo calibro Granulomatosi di Wegener Sindrome di Churg – Strauss Poliangioite microscopica Vasi di piccolo calibro Crioglobulinemia mista essenziale Angioite cutanea leucocitoclasica Porpora di Schönlein – Henoch Vasculite orticarioide Porpora ipergammaglobulinemica benigna Eritema elevatum diutinum
 Sclerolupus (Sclerosi sistemica – Lupus eritematoso sistemico) Sclerosi sistemica – Cirrosi biliare primitiva – Sindrome di Sjögren. Altre sindromi da sovrapposizione Connettiviti indifferenziate 2.7. Vasculiti sistemiche (Primitive) Arterie di grosso e medio calibro Arterite di Takayasu. Arterite gigantocellulare di Horton. Arterie di medio calibro Panarterite nodosa. Vasi di medio e piccolo calibro. Granulomatosi di Wegener. Sindrome di Churg – Strauss. Poliangioite microscopica. Vasi di piccolo calibro. Crioglobulinemia mista essenziale. Angioite cutanea leucocitoclasica. Porpora di Schönlein – Henoch. Vasculite orticarioide. Porpora ipergammaglobulinemica benigna. Eritema elevatum diutinum.")
7
Altre vasculiti Malattia di Behçet Malattia di Kawasaki
Malattia di Behçet Malattia di Kawasaki Sindrome di Cogan Vasculite linfocitaria benigna Vasculiti overlap o non classificabili Vasculiti sistemiche (Secondarie) A farmaci e tossici Ad agenti infettivi e vaccini Associate ad altre condizioni Artrite reumatoide Connettiviti Neoplasie Pseudovasculiti 2.8. Sindrome da anticorpi antifosfolipidi Primitiva Associata ad altre malattie 2.9. Polimialgia reumatica Associata ad arterite gigantocellulare di Horton 2.10. Eritema nodoso 2.11 Panniculiti 2.12. Policondriti
 A farmaci e tossici Ad agenti infettivi e vaccini. Associate ad altre condizioni. Artrite reumatoide. Connettiviti. Neoplasie. Pseudovasculiti 2.8. Sindrome da anticorpi antifosfolipidi. Primitiva. Associata ad altre malattie 2.9. Polimialgia reumatica. Associata ad arterite gigantocellulare di Horton Eritema nodoso Panniculiti Policondriti.")
8
per il riconoscimento precoce delle “connettiviti”.
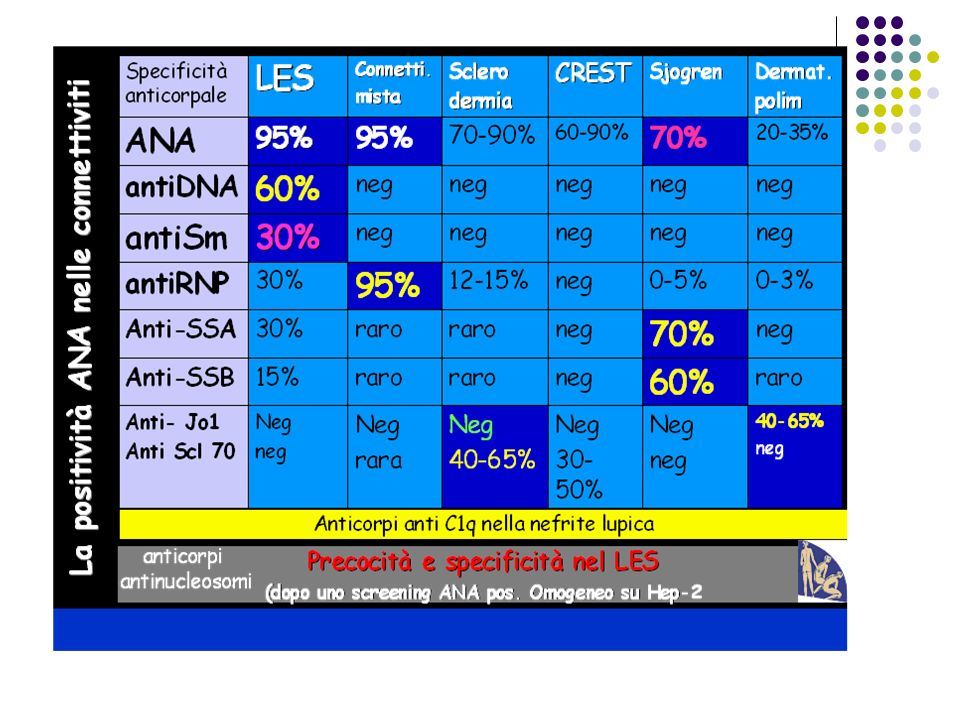
Dalla complessità di tale classificazione si evince la grossa difficoltà per il riconoscimento precoce delle “connettiviti”. Il medico di medicina generale può comunque sospettare la presenza di una connettivite quando si trovino determinati dati del laboratorio generale, più mirati e più specifici per questo gruppo di malattie,che consentano di confermare una diagnosi sospetta,di determinarne la gravità, di sorvegliare il decorso della malattia, di valutare la efficacia della terapia e, in molti casi, di studiare in via prospettica la prognosi o la possibile comparsa di temibili complicanze a carico di qualsiasi organo (polmone, rene,vasi,etc). E’ possibile, infatti, sfruttando una griglia di parametri di laboratorio "specifici" pervenire ad una diagnosi abbastanza precisa del tipo di malattia.
. E’ possibile, infatti, sfruttando una griglia di parametri di laboratorio specifici pervenire ad una diagnosi abbastanza precisa del tipo di malattia.")
9
Le connettiviti sono malattie acquisite ad eziologia autoimmune del tessuto connettivo, che hanno alla base la caratteristica di aver perso la tolleranza immunologica verso qualche antigene self. Le forme principali di connettiviti sono: • Artrite reumatoide (articolazioni) • S. di Sjogren (ghiandole esocrine) • Sclerosi sistemica (cute) • Polimiosite (muscoli) • Connettivite mista • LES
 • S. di Sjogren (ghiandole esocrine) • Sclerosi sistemica (cute) • Polimiosite (muscoli) • Connettivite mista. • LES.")
10
Tutte queste forme di malattia hanno alcuni aspetti in comune:
- Clinici: o artriti e artralgie o febbre o lesioni cutanee di vario tipo o fenomeno di Raynaud - Laboratoristici: o iperglobulinemia o aumento della VES o anemia non emolitica o leucopenia o diminuzione della proteidemia complementare o Lupus Band Test positivo (immunofluorescenza di biopsia cutanea che evidenzia immunocomplessi alla giunzione dermo-epidermica) o Fattore reumatoide o Ab anti nucleo
 o Fattore reumatoide. o Ab anti nucleo.")
11
“CONOSCI IL LUPUS, CONOSCI LA MEDICINA INTERNA”
Per molti anni è stata opinione comune che i cosiddetti “reumatismi” interessassero quasi esclusivamente le articolazioni. Solo negli ultimi anni, grazie anche a una maggiore conoscenza dei meccanismi eziopatogenetici e immunopatologici che sono alla base dell’insorgenza e dello sviluppo delle patologie del connettivo, la Reumatologia ha visto riconosciuto un ruolo centrale nell’ambito della Medicina Interna.

12
Diagnosi e laboratorio
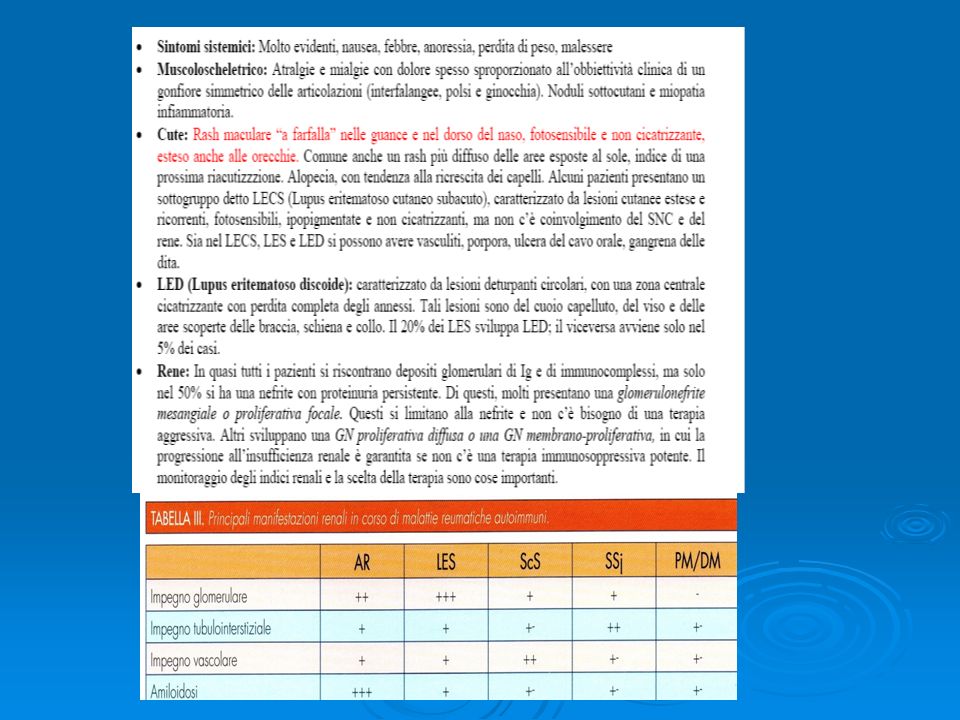
- ANA (anti nuclear antiobodies) sono il parametro più affidabile presente nel 95% dei malati con lupus ma non sono specifici. Relativamente specifici sono invece gli anticorpi contro il DNA a doppia elica (ds-DNA) ANA + dsDNA + complementemia ridotta indicano malattia in fase attiva. - CH50 (test di attivazione del complemento) è la misura più specifica dell’attività complementare ma è soggetto a molti errori di laboratorio. Per la diagnosi di LES devono essere soddisfatti almeno 4 degli 11 criteri riportati qui di seguito: 1. Rash malare 2. Lupus discoide 3. Fotosensibilità 4. Ulcere al cavo orale 5. Artrite 6. Sierositi 7. Nefropatia 8. Danno neurologico 9. Alterazioni ematologiche 10. Disordini immunologici 11. Anticorpi antinucleo
 sono il parametro più affidabile presente nel 95% dei malati con lupus ma non sono specifici. Relativamente specifici sono invece gli anticorpi contro il DNA a doppia elica (ds-DNA) ANA + dsDNA + complementemia ridotta indicano malattia in fase attiva. - CH50 (test di attivazione del complemento) è la misura più specifica dell’attività complementare ma è soggetto a molti errori di laboratorio. Per la diagnosi di LES devono essere soddisfatti almeno 4 degli 11 criteri riportati qui di seguito: 1. Rash malare. 2. Lupus discoide. 3. Fotosensibilità. 4. Ulcere al cavo orale. 5. Artrite. 6. Sierositi. 7. Nefropatia. 8. Danno neurologico. 9. Alterazioni ematologiche. 10. Disordini immunologici. 11. Anticorpi antinucleo.")
18
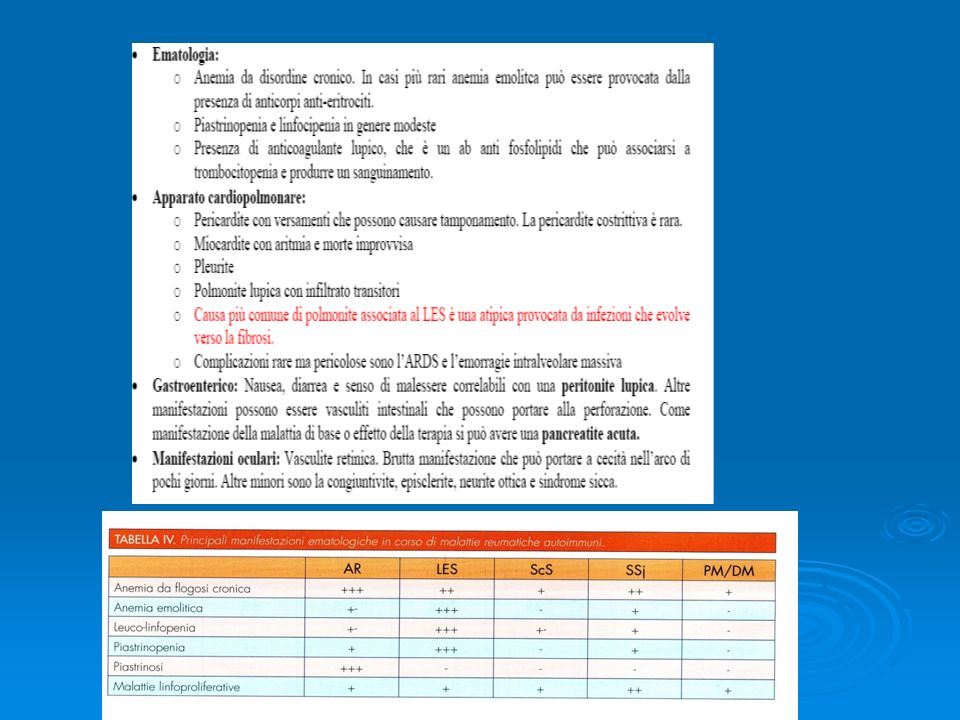
SNC:Tutti i distretti possono essere interessati,sia le meningi
che i nervi,che il midollo spinale Sistema vascolare: una delle complicanze piu’ temuta è il verificarsi di trombosi in ogni distretto. Invece che alle vasculiti sembrano legate all’associazione fra anticorpi antifosfolipidi e processi coagulativi. Inoltre le continue alterazioni endoteliali associate alla deposizione degli immunocomplessi e l’iperlipidemia secondaria alla terapia steroidea possono provocare coronaropatia con angina instabile

19
DERMATOMIOSITE E POLIMIOSITE
Malattie a presunta eziologia autoimmune che provocano un danneggiamento del muscolo scheletrico attraverso una infiammazione non suppurativa con infiltrato linfocitario. Polimiosite si ha quando viene risparmiata la cute, mentre nella dermatomiosite c’è anche un caratteristico rash cutaneo. Un terzo di questi casi si associa ad altre connettiviti, un decimo a neoplasie maligne. L’eziologia più probabile sembra una reattività crociata, legata ad aplotipi HLA DR3 e DRw52, di proteine muscolari ed antigeni del virus Coxsackie, capace già da solo di dare alcuni quadri di miosite. Di importanza rilevante è sicuramente la presenza di un danno vascolare mediato dai CD8 che produce e precede la distruzione del tessuto muscolare. Ci sono 5 tipi di malattia: 1. Polimiosite idiopatica primitiva 2. Dermatomiosite idiopatica primitiva 3. Dermatomiosite (polimiosite) associata a neoplasia 4. Dermatomiosite (polimiosite) associata a vasculite (infantile) 5. Dermatomiosite (polimiosite) associata a collagenopatia
 associata a neoplasia. 4. Dermatomiosite (polimiosite) associata a vasculite (infantile) 5. Dermatomiosite (polimiosite) associata a collagenopatia.")
20
SINDROME DI SJOGREN Malattia autoimmune a decorso lentamente progressivo che colpisce con una infiltrazione linfocitaria le ghiandole esocrine, e determina xerostomia (bocca secca) e secchezza oculare. Un terzo dei pazienti ha anche sintomi extraghiandolari. La malattia può essere primitiva o associata ad altre patologie reumatologiche autoimmuni. Colpisce le donne di mezza età (F>M 9:1) Clinica La principale manifestazione è la mancanza di secrezione oculare e salivare, che provoca la secchezza di questi organi. A questo si associano una serie di manifestazioni legate all’interessamento di altre ghiandole e di siti extraghiandolari. Infine sono presenti dei sintomi sistemici.
 e secchezza oculare. Un terzo dei pazienti ha anche sintomi extraghiandolari. La malattia può essere primitiva o associata ad altre patologie reumatologiche autoimmuni. Colpisce le donne di mezza età (F>M 9:1) Clinica. La principale manifestazione è la mancanza di secrezione oculare e salivare, che provoca la secchezza di questi organi. A questo si associano una serie di manifestazioni legate all’interessamento di altre ghiandole e di siti extraghiandolari. Infine sono presenti dei sintomi sistemici.")
21
SCLERODERMIA (SCLEROSI SISTEMICA)
Malattia autoimmune molto variabile, caratterizzata da una fibrosi progressiva dalla cute, dei vasi sanguigni e di organi come il polmone, il rene, il tubo digerente e il cuore. La gravità delle lesioni e l’interessamento sistemico variano nei pazienti. Esistono comunque due grandi raggruppamenti clinici, quello della sclerodermia cutanea diffusa, che si manifesta con ispessimento cutaneo di tutti i distretti e un interessamento viscerale più grave, e la sclerodermia cutanea limitata, con ispessimento della cute delle estremità e del volto e prognosi più favorevole (cioè CREST: Calcinosi, fenomeno di Raynaud, Esofago ipomobile, Sclerodattilia e Teleangectasia). Infine c’è la sclerosi sistemica senza interessamento cutaneo. La sopravvivenza è legata alla gravità delle manifestazioni a carico del cuore, dei reni e del polmone. La sclerosi sistemica può manifestarsi anche in associazione con le manifestazioni di altre connettiviti. Questi quadri clinici sono stati denominati Overlap Syndrome.
. Infine c’è la sclerosi sistemica senza interessamento cutaneo. La sopravvivenza è legata alla gravità delle manifestazioni a carico del cuore, dei reni e del polmone. La sclerosi sistemica può manifestarsi anche in associazione con le manifestazioni di altre connettiviti. Questi quadri clinici sono stati denominati Overlap Syndrome.")
22
SCLERODERMIA Sono le manifestazioni vascolari, evento precoce che si manifesta con il fenomeno di Raynauld, che producono l’attivazione dei fibroblasti. Queste interessano prima di tutto le arterie di piccolo calibro, le arteriole e i capillari cutanei degli organi interessati dai processi patologici. Danno della cellula endoteliale ispessimento dell’intima ostruzione vasale stato di ischemia cronica dell’organo interessato. Il danno vascolare può anche essere osservato nei capillari cutanei con la capillarografia.

23
CONNETTIVITE MISTA Malattia caratterizzata dall’associazione di manifestazioni del LES, della sclerodermia, della polimiosite e dell’artrite reumatoide. La presenza di anticorpi anti RNP plasmatici permette di catalogarla come una malattia a se’. Insorge preferenzialmente nella seconda-terza decade e colpisce di più le donne. I meccanismi patogenetici sono quelli delle malattie da cui deriva.

24
Esordio classico: fenomeno di Raynaud, mani gonfie, artralgia, mialgia e astenia. Le varie manifestazioni che completano il quadro si sviluppano nel corso di mesi o anni. • Tumefazione delle dita con possibile evoluzione verso la sclerodattilia. • Alterazioni di tipo sclerodermico della cute delle estremità distali e del volto, mai del tronco • Alcuni rash maculare (a volte discoide) e fotosensibilità • Dolori, rigidità e tumefazioni delle articolazioni periferiche • Deformità delle mani come nell’AR ma senza erosione ossea • Lesioni muscolari tipiche della polimiosite, con alterazioni dell’EMG e aumento degli enzimi muscolari • 85% dei pazienti presenta interessamento polmonare in genere asintomatico. Abbastanza comune la pleurite. • 25% presenta glomerulopatia membranosa in genere molto lieve. • 70% manifestazioni gastroenteriche, alterazioni dello sfintere esofageo e reflusso gastrico • 30% pericardite e disturbi del cuore • Anemia da disordine cronico • Test di Coombs + nel 60% dei pazienti ma rara l’anemia emolitica • Ipergammaglobulinemia e fattore reumatoide frequenti • Anticorpi anti U1 RNP, specifici per una proteina di 70KD legata all’RNA a basso peso molecolare.
 e fotosensibilità. • Dolori, rigidità e tumefazioni delle articolazioni periferiche. • Deformità delle mani come nell’AR ma senza erosione ossea. • Lesioni muscolari tipiche della polimiosite, con alterazioni dell’EMG e aumento degli enzimi muscolari. • 85% dei pazienti presenta interessamento polmonare in genere asintomatico. Abbastanza comune la pleurite. • 25% presenta glomerulopatia membranosa in genere molto lieve. • 70% manifestazioni gastroenteriche, alterazioni dello sfintere esofageo e reflusso gastrico. • 30% pericardite e disturbi del cuore. • Anemia da disordine cronico. • Test di Coombs + nel 60% dei pazienti ma rara l’anemia emolitica. • Ipergammaglobulinemia e fattore reumatoide frequenti. • Anticorpi anti U1 RNP, specifici per una proteina di 70KD legata all’RNA a basso peso molecolare.")
25
La Sindrome da anticorpi antifosfolipidi (APS) è un disordine acquisito
di origine ignota, caratterizzato da trombosi arteriose e/o venose e complicanze della gravidanza che si associano alla presenza nel sangue degli anticorpi antifosfolipidi (aPL) (1). Gli aPL allungano i tempi di coagulazione dei tests fosfolipide-dipendenti della coagulazione (si parla in questo caso di anticoagulante tipo lupus, LAC) (2), oppure sono evidenziati mediante metodiche ELISA che utilizzano la cardiolipina o altri fosfolipidi a carica netta negativa come antigeni in fase solida (anticorpi anticardiolipina, aCL) (3). In realtà, gli aPL non reagiscono direttamente con i fosfolipidi, bensì sono diretti contro proteine plasmatiche che hanno affinità per le superfici a carica netta negativa. Tra queste proteine, le più importanti sono la ß2-glicoproteina 1 (ß2GP1)(4) e la protrombina (PT) (5), che sono il bersaglio antigenico della maggior parte degli aPL.
 (1). Gli aPL allungano i tempi di coagulazione dei tests fosfolipide-dipendenti. della coagulazione (si parla in questo caso di anticoagulante tipo lupus, LAC) (2), oppure sono evidenziati mediante metodiche ELISA che utilizzano. la cardiolipina o altri fosfolipidi a carica netta negativa come antigeni in fase. solida (anticorpi anticardiolipina, aCL) (3). In realtà, gli aPL non reagiscono direttamente con i fosfolipidi, bensì sono. diretti contro proteine plasmatiche che hanno affinità per le superfici a carica. netta negativa. Tra queste proteine, le più importanti sono la ß2-glicoproteina. 1 (ß2GP1)(4) e la protrombina (PT) (5), che sono il bersaglio antigenico. della maggior parte degli aPL.")
26
SINDROME DA ANTICORPI ANTIFOSFOLIPIDI
Nel tempo la sindrome si è dunque evoluta come concetto di patologia autoimmune che riguarda equilibri immunologici dell’intero organismo, non relativi solo ai meccanismi che regolano la coagulazione. L’insieme delle manifestazioni correlabili con la sindrome è cambiato e si sono studiati possibili legami esistenti con espressioni neurologiche di natura apparentemente diversa (somiglianti a quelle ad esempio della sclerosi multipla, dell’epilessia ecc), con alterazioni aterosclerotiche precoci. Esistono inoltre coinvolgimenti noti delle valvole cardiache, della pelle (livedo reticularis, ulcerazioni), ematologici (diminuito numero di piastrine nel sangue) e sintomi specifici differenti in rapporto agli organi coinvolti (reni, cuore, cervello, arti, ecc)
, con alterazioni aterosclerotiche precoci. Esistono inoltre coinvolgimenti noti delle valvole cardiache, della pelle (livedo reticularis, ulcerazioni), ematologici (diminuito numero di piastrine nel sangue) e sintomi specifici differenti in rapporto agli organi coinvolti (reni, cuore, cervello, arti, ecc)")
27
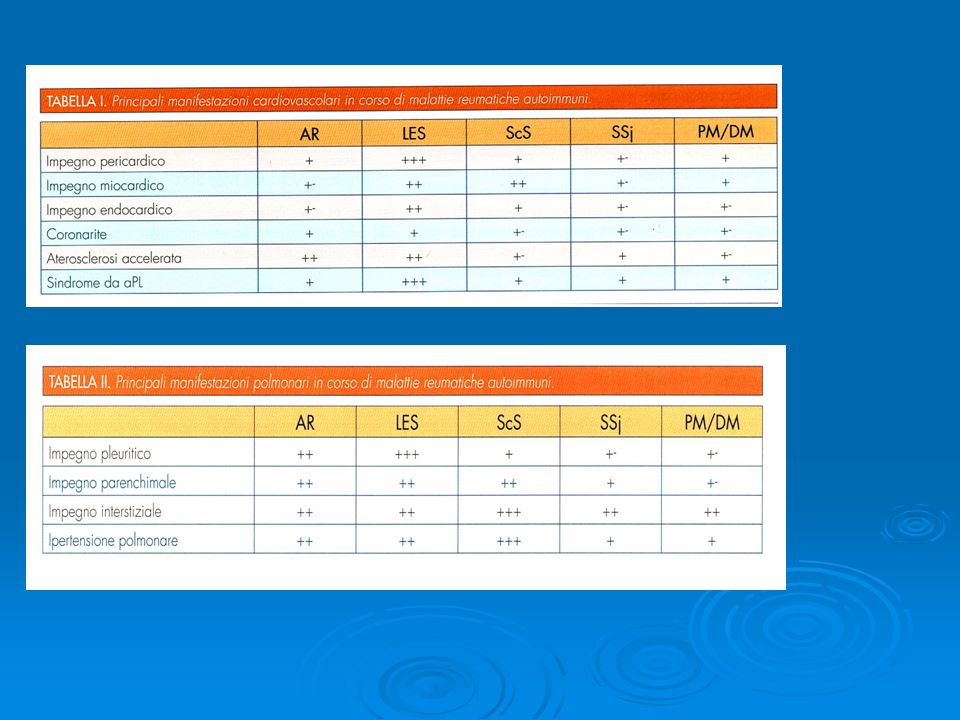
Le Vasculiti sono caratterizzate da un processo infiammatorio che interessa la parete dei vasi sanguigni che conduce ad alterazioni del flusso ematico e a danno dell'integrità del vaso. I vasi coinvolti possono essere di diverso tipo o calibro con conseguenze su uno o più organi o apparati. Le sindromi cliniche che ne derivano sono per lo più conseguenza dell'ischemia tissutale, del danno vasale e dell'infiammazione sistemica con febbre, anoressia e calo ponderale La distinzione tra vasculiti PRIMITIVE e SECONDARIE nasce dal fatto che queste ultime sono coesistenti a malattie ben caratterizzate da un punto di vista patogenetico e sierologico mentre le prime non hanno alcuna di queste caratteristiche. Alla luce di questa definizione rimangono numerosi dubbi se alcune vasculiti fino ad ora considerate primitive come la GW non debbano, alla luce della caratterizzazione degli ANCA, essere considerate secondarie. Comunque, nella maggioranza dei casi, un test specifico per la diagnosi di vasculite manca e la diagnosi deve ancora essere basata sulla dimensione dei vasi interessati e sulla presenza di quadri clinici e di laboratorio assolutamente non specifici.

28
VASCULITI -dei vasi di grosso calibro: dei vasi di medio calibro: Arterite di Horton Poliarterite nodosa Arterite di Takayasu M.di Kawasaki(bambini) -dei vasi di piccolo calibro: ANCA anticorpi diretti contro componenti citoplasmatici dei granulociti neutrofili Anca + Micropoliarterite Granulomatosi di Wegener Malattia di Churc Strauss Anca - Crioglobulinemia mista Porpora di Schonlein-Henoch MISCELLANEA: -M.di Burger -M.di Behcet Anche il siero di pazienti con altre vasculiti (ma anche altre malattie) può contenere ANCA ma in genere in un pattern perinucleare(P-ANCA). Infatti il 50% dei casi di MP sono C-ANCA positivi ma il rimanente è positivo per i P-ANCA. La specificità antigenica di questi ultimi è costituita dalla mieloperossidasi, un altro enzima contenuto all'interno dei granuli dei neutrofili. Dal momento che i P-ANCA possono essere positivi in numerose altre patologie anche non vasculitiche come la colite ulcerosa, l'artrite reumatoide, la malattia di Crohn, l'epatite autoimmune, il valore predittivo positivo di questo pattern è più basso di quello dei C-ANCA.
 -dei vasi di piccolo calibro: ANCA anticorpi diretti contro componenti citoplasmatici. dei granulociti neutrofili. Anca + Micropoliarterite Granulomatosi di Wegener. Malattia di Churc Strauss. Anca - Crioglobulinemia mista Porpora di Schonlein-Henoch. MISCELLANEA: -M.di Burger. -M.di Behcet. Anche il siero di pazienti con altre vasculiti (ma anche altre malattie) può contenere ANCA ma in genere in un pattern perinucleare(P-ANCA). Infatti il 50% dei casi di MP sono C-ANCA positivi ma il rimanente è positivo per i P-ANCA. La specificità antigenica di questi ultimi è costituita dalla mieloperossidasi, un altro enzima contenuto all interno dei granuli dei neutrofili. Dal momento che i P-ANCA possono essere positivi in numerose altre patologie anche non vasculitiche come la colite ulcerosa, l artrite reumatoide, la malattia di Crohn, l epatite autoimmune, il valore predittivo positivo di questo pattern è più basso di quello dei C-ANCA.")
29
Manifestazioni cliniche delle vasculiti
Sintomi generali: febbre, astenia, calo ponderale, ipertensione arteriosa. La febbre può essere continua, remittente o intermittente, può esserci febbricola o temperatura elevata con caratteri da far pensare ad una infezione o ad una sepsi, per cui spesso vengono allestiti esami colturali. Apparato muscolo-scheletrico: artriti, artralgie, mialgie. Cute: noduli, ulcere, gangrene distali, porpora, eritema nodoso o multiforme, orticaria, livedo reticularis. Rene: proteinuria, ematuria (spesso microscopica), insufficienza renale. Cuore: cardiomegalia, angina, infarto, scompenso. Apparato digerente: dolori addominali, nausea, vomito, segni di ischemia intestinale, perforazione. Sistema Nervoso Centrale: disturbi psichici, cefalea, convulsioni, accidenti cerebrovascolari. Sistema Nervoso Periferico: neuropatie sensitive e/o motorie, mononeurite multipla. Altri organi: occhi (uveite, irite, iridociclite, ischemia o emorragia retiniche), testicoli (PAN classica), polmoni (Malattia di Wegener e Malattia di Churg-Strauss).
, insufficienza renale. Cuore: cardiomegalia, angina, infarto, scompenso. Apparato digerente: dolori addominali, nausea, vomito, segni di ischemia intestinale, perforazione. Sistema Nervoso Centrale: disturbi psichici, cefalea, convulsioni, accidenti cerebrovascolari. Sistema Nervoso Periferico: neuropatie sensitive e/o motorie, mononeurite multipla. Altri organi: occhi (uveite, irite, iridociclite, ischemia o emorragia retiniche), testicoli (PAN classica), polmoni (Malattia di Wegener e Malattia di Churg-Strauss).")
30
livelli specialistici piu’ idonei all’inquadramento nosografico.
CONCLUSIONI Rimane comunque saldo il concetto del precoce riconoscimento delle Connettiviti per una più efficace e corretta “Clinical governance” del paziente affetto da tali patologie ed il MMG rimane il primo”Filtro”essenziale,nel sospetto,per l’invio ai livelli specialistici piu’ idonei all’inquadramento nosografico.

34
“IL MMG: APPROCCIO ALLE CONNETTIVITI ”
L’approccio alle CONNETTIVITI rappresenta un ulteriore e forse più complesso “problem solving” per il Medico di Medicina Generale rispetto alle artriti trattate nel precedente I° Seminario,in quanto ancora più variegato il corteo sintomatologico ed il più delle volte di difficile interpretazione in diagnosi differenziale.

Presentazioni simili