Scaricare la presentazione
1
Malattia di Parkinson (M. Fasano – R. Fesce)
")
2
Malattia di Parkinson “Involuntary, tremulous motion, with lessened muscular power, in parts not in action and even when supported; with a propensity to bend the trunk forwards, and to pass from a walking to a running pace: the senses and intellects being uninjured.” James Parkinson, Essay on the Shaking Palsy, London 1817

3
Malattia di Parkinson Seconda malattia neurodegenerativa più comune dopo Alzheimer Prevalenza: 1-2% fino a 65 anni 4-5% oltre 65 Esordio generalmente tardivo: “Late-onset Parkinson’s Disease” “Early-onset Parkinson’s Disease” <50 anni

4
Segni clinici e patologici
Tremore a riposo, unilaterale all’esordio Bradicinesia Rigidità Corpi di inclusione intracellulari eosinofili (corpi di Lewy) Depigmentazione della substantia nigra pars compacta Lewy FH (1912) Paralysis agitans. Pathologische Anatomie. In: Handbuch der Neurologie Band III (Lewandowsky M, Abelsdorff G, eds), pp Berlin: Springer Verlag.
 Depigmentazione della substantia nigra pars compacta. Lewy FH (1912) Paralysis agitans. Pathologische Anatomie. In: Handbuch der Neurologie Band III (Lewandowsky M, Abelsdorff G, eds), pp Berlin: Springer Verlag.")
5
La substantia nigra pars compacta e la via nigrostriatale
Normale Parkinson

6
Anatomia funzionale dei nuclei della base
Blandini et al., 2000

7
Genetic Parkinsonisms (10-15%)
PARK1: AD α-synuclein (mut.) PARK2: AR parkin PARK3: AD, 2p13 PARK4: AD Iowa kindred α-synuclein (gene dupl./tripl.) PARK5: AD? UCH-L1 PARK6: AR PINK1 PARK7: AR DJ-1 PARK8: AD LRRK2, dardarin PARK9: AR ATP13A2 PARK10: non-Mendelian, 1p32 PARK11: AD GIGYF2 PARK12: non-Mendelian, Xq PARK13: non-Mendelian OMI/HtrA2 PARK14: AR PLA2G6 PARK15: AR FBXO7 Lesage and Brice, Human Molecular Genetics 2009
 PARK2: AR parkin. PARK3: AD, 2p13. PARK4: AD Iowa kindred α-synuclein (gene dupl./tripl.) PARK5: AD UCH-L1. PARK6: AR PINK1. PARK7: AR DJ-1. PARK8: AD LRRK2, dardarin. PARK9: AR ATP13A2. PARK10: non-Mendelian, 1p32. PARK11: AD GIGYF2. PARK12: non-Mendelian, Xq. PARK13: non-Mendelian OMI/HtrA2. PARK14: AR PLA2G6. PARK15: AR FBXO7. Lesage and Brice, Human Molecular Genetics")
8
Meccanismi patogenetici convergenti
Fasano, Alberio & Lopiano, Biomarkers Med. 2, (2008)
")
9
Principale componente dei corpi di Lewy
α-Sinucleina: Principale componente dei corpi di Lewy 1912: Friedrich Lewy descrisse i corpi di inclusione per la prima volta 1998: l’α-sinucleina è la principale componente proteica dei corpi di Lewy insieme a proteine ubiquitinilate M.G. Spillantini et al., PNAS 95, (1998)
")
11
Cosa determina l’aggregazione dell’α-sinucleina?
α-syn è overespressa (PARK4) α-syn non viene degradata (proteasomal impairment) α-syn è modificata da prodotti di ossidazione della dopamina
 α-syn non viene degradata (proteasomal impairment) α-syn è modificata da prodotti di ossidazione della dopamina.")
12
Interazione tra α-Syn e dopamina
Halliday et al, Brain 2005 Conway et al., Science 2001 Lotharius & Brundin, Hum. Mol. Genet. 2002 Fasano et al., Neurochem. Int. 2003 Bisaglia et al, Biochemistry 2005

13
Parkina

14
La via Ubiquitina-Proteasoma
Brazil, Nat Rev Neurosci 4, 698 (2003) Cook and Petrucelli, BBA Mol Basis Dis 2009
 Cook and Petrucelli, BBA Mol Basis Dis")
15
DJ-1 Bonifati V, Oostra BA, Heutink P. J Mol Med. 2004; 82:

16
DJ-1 protegge i neuroni dalla tossicità della dopamina

17
In the PINK… Welberg, Nat Rev Neurosci 9, 167 (2008)
Fitzgerald & Plun-Favreau, FEBS J 275, (2008)
")
18
Tan JM, Dawson TM. Parkin blushed by PINK1. Neuron. 2006; 50:527-529.

19
Possibili ruoli neuroprotettivi per PINK1
A model of the putative neuroprotective mechanisms of PINK1. PINK1 may maintain neuronal survival by phosphorylating specific mitochondrial proteins or binding to key signaling molecules in signaling pathways governing cell death and survival. Upon phosphorylation, these proteins may mediate neuroprotective signals of PINK1 by four major mechanisms. Mechanism 1: PINK1 modulates the E3 ligase activity of Parkin. Parkin then exerts neuroprotective action by inducing Lys63 polyubiquitination and activation of IKK complex. The activated IKK phosphorylates IκB, relieving its inhibition of NF-κB. The uninhibited NF-κB enters the nucleus and directs transcription of genes that enhance neuronal survival. Parkin also enhances phosphorylation and activation of Akt by an unknown mechanism. The activated Akt then suppresses the action of the pro-apoptotic JNK pathway and block Bax to induce release of cytochrome c and other pro-apoptotic proteins from mitochondria. Mechanism 2: PINK1 facilitates Bcl-2 inhibition of the release of the mitochondrial proteins involved in regulating cell death such as cytochrome c and endonuclease G (EndoG). Mechanism 3: PINK1 binds and phosphorylates TRAP1. Upon phosphorylation, TRAP1 is activated and blocks generation of mitochondrial ROS. As ROS induces opening of mitochondrial permeability transition pores (PTPs), PINK1, and TRAP1 can therefore indirectly block this opening and hence prevent release of cytochrome c and other pro-apoptotic proteins from mitochondria. Mechanism 4: PINK1 enhances phosphorylation and activation of the mitochondrial protease HtrA2 by the stress activated MAP kinase p38γ. Oxidative stress induces phosphorylation and activation of p38γ by upstream kinases such as MEKK3. By binding to HtrA2, PINK1 enhances phosphorylation of Ser142 of HtrA2 by p38γ. The phosphorylated HtrA2 then protects neuronal cells from undergoing apoptosis induced by neurotoxins and oxidative stress. Mills et al., J Neurochem 105, (2008)
. Mechanism 3: PINK1 binds and phosphorylates TRAP1. Upon phosphorylation, TRAP1 is activated and blocks generation of mitochondrial ROS. As ROS induces opening of mitochondrial permeability transition pores (PTPs), PINK1, and TRAP1 can therefore indirectly block this opening and hence prevent release of cytochrome c and other pro-apoptotic proteins from mitochondria. Mechanism 4: PINK1 enhances phosphorylation and activation of the mitochondrial protease HtrA2 by the stress activated MAP kinase p38γ. Oxidative stress induces phosphorylation and activation of p38γ by upstream kinases such as MEKK3. By binding to HtrA2, PINK1 enhances phosphorylation of Ser142 of HtrA2 by p38γ. The phosphorylated HtrA2 then protects neuronal cells from undergoing apoptosis induced by neurotoxins and oxidative stress. Mills et al., J Neurochem 105, (2008)")
20
Parkinson, malattia mitocondriale?
Henchcliffe and Beal, Nature Clinical Practice Neurology 4, (2008)
")
21
Dardarina, LRRK2

22
L’ipotesi di Braak (2004)
")
23
Un possibile quadro d’unione

24
Meccanismi patogenetici convergenti
Yang YX, Wood NW, Latchman DS. Neuroreport. 2009; 20:

25
Alterazioni dell’omeostasi della dopamina (DA)
H2O2 Specie reattive dell’ossigeno Aime, Bergamasco, Casu, Digilio, Fasano, Giraudo, Lopiano, Movement Disorders, 2000. Fasano & Lopiano, 2008

26
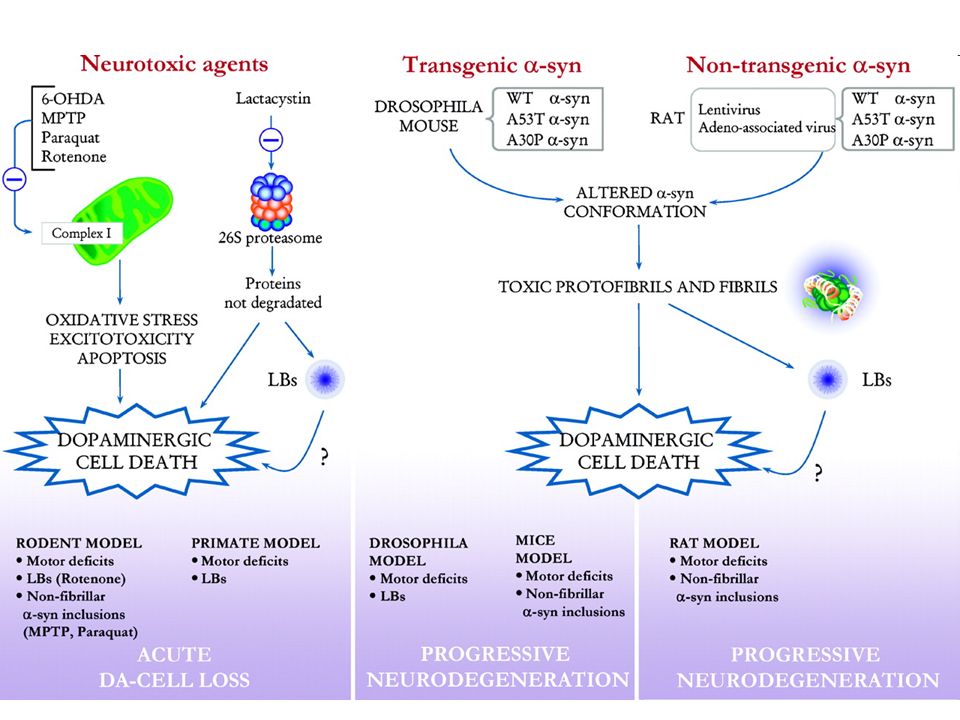
Modelli sperimentali Neurotossine Cellulari Animali Genetici MPTP
Rotenone 6-OHDA MDMA Genetici Cellulari Animali

27
I “frozen addicts” di J.W. Langston: La scoperta di MPTP
Vila M, Przedborski S. Nat Rev Neurosci. 2003; 4:

28
Il modello Rotenone Adam, Nature 408, 125 (2000)
")
29
Alterata omeostasi della dopamina: il modello SH-SY5Y
↑DAT ↓VMAT

30
Terapie neuroprotettive possibili?
Dawson e Dawson, 2003

31
Terapie neuroprotettive possibili?
Dawson e Dawson, 2003

32
Terapie neuroprotettive possibili?

33
Terapie neuroprotettive possibili?
Manca un controllo dell’efficacia (outcome measure) La diagnosi tardiva rende inefficaci le terapie neuroprotettive
 La diagnosi tardiva rende inefficaci le terapie neuroprotettive.")
34
due input paralleli: cortico-striatale e cortico-subtalamico
circuiti di feedback interno che regolano output dei NB (attraverso GPI e SNr) DA privilegia ritorno facilitatorio “diretto” (cortico-putamino-GPI) su ritorno inibitorio (cortico-putamino-GPE-[STN]-GPI) interneuroni colinergici essenzialmente inibiti da DA Parkinson – ritorno specifico sbilanciato verso inibizione (acinesia, bradicinesia, riduzione movimenti involontari) – generica inibizione della corteccia con minor controllo corticale su circuiti e riflessi sottocorticali (rigidità, tremore) importanza della circuiteria mesencefalica per gratificazione e motivazione: contributo a depressione, rallentamento motorio e cognitivo in PD
 DA privilegia ritorno facilitatorio diretto (cortico-putamino-GPI) su ritorno inibitorio (cortico-putamino-GPE-[STN]-GPI) interneuroni colinergici essenzialmente inibiti da DA. Parkinson – ritorno specifico sbilanciato verso inibizione (acinesia, bradicinesia, riduzione movimenti involontari) – generica inibizione della corteccia con minor controllo corticale su circuiti e riflessi sottocorticali (rigidità, tremore) importanza della circuiteria mesencefalica per gratificazione e motivazione: contributo a depressione, rallentamento motorio e cognitivo in PD.")
35
armamentario terapeutico tradizionale
agonisti DA-ergici (apomorfina, bromocriptina, pramipexolo) l-DOPA inibitori decarbossilasi periferica (carbiDOPA, benserazide) antimuscarinici (benztropina, triesifenidile) Nessun lavoro rilevante negli ultimi 10 anni: attivi soprattutto su tremore, invertono iperattività colinergica da DA. Limitati da effetti anticolinergici (periferici e) centrali: sedazione, allucinazioni, perdita di memoria amantadina ([antivirale] stimolante turnover DA ?) Blanda efficacia, effetti collaterali anticolinergici, ed altri. Aumenta rilascio e riduce reuptake di DA(?). Forse può anche legare il recettore difetti della terapia l-DOPA unico veramente efficace trattamento con l-DOPA sembrava limitato nel tempo poi subentra oscillazione tra discinesie (ON drug) e acinesia (OFF drug)
 l-DOPA. inibitori decarbossilasi periferica (carbiDOPA, benserazide) antimuscarinici (benztropina, triesifenidile) Nessun lavoro rilevante negli ultimi 10 anni: attivi soprattutto su tremore, invertono iperattività colinergica da DA. Limitati da effetti anticolinergici (periferici e) centrali: sedazione, allucinazioni, perdita di memoria. amantadina ([antivirale] stimolante turnover DA ) Blanda efficacia, effetti collaterali anticolinergici, ed altri. Aumenta rilascio e riduce reuptake di DA( ). Forse può anche legare il recettore. difetti della terapia. l-DOPA unico veramente efficace. trattamento con l-DOPA sembrava limitato nel tempo. poi subentra oscillazione tra discinesie (ON drug) e acinesia (OFF drug)")
36
perché l-DOPA da sola non va bene?
problemi essenzialmente farmacocinetici biodisponibilità cerebrale inibiz. DOPA-decarbox (non BEE permeant) t1/2 breve (1,5 h): a lungo termine effetto rispecchia livello plasmatico si punta a CDS: Continuous DA-ergic Stimulation agonisti DA a durata più lunga derivati dell’ergot: bromocriptina, cabergolina, di-H-ergocriptina, pergolide non-ergot: piribedil, pramipexolo, ropinirolo [apomorfina e lisuride OK per sé, ma hanno durata più breve] rotigotina [agonista D2 lipofilo per somministraz. transdermica] inibizione MAO-B pargilina, selegilina (bene preparaz. boccale) rasargilina (nuovo selettivo irreversibile) inibizione COMT entacapone, tolcapone (epatotossico) [fluttuazioni plasmatiche di l-D ridotte del 30-40%. ON-time giornaliero +1-2 ore. Dose giorn. di l-D –10-30%] preparazioni a rilascio lento, transdermiche, sottocutanee, nanoparticles lipidiche solide e microparticles biocompatibili infusione continua digiunale
 t1/2 breve (1,5 h): a lungo termine effetto rispecchia livello plasmatico. si punta a CDS: Continuous DA-ergic Stimulation. agonisti DA a durata più lunga derivati dell’ergot: bromocriptina, cabergolina, di-H-ergocriptina, pergolide non-ergot: piribedil, pramipexolo, ropinirolo [apomorfina e lisuride OK per sé, ma hanno durata più breve] rotigotina [agonista D2 lipofilo per somministraz. transdermica] inibizione MAO-B pargilina, selegilina (bene preparaz. boccale) rasargilina (nuovo selettivo irreversibile) inibizione COMT entacapone, tolcapone (epatotossico) [fluttuazioni plasmatiche di l-D ridotte del 30-40%. ON-time giornaliero +1-2 ore. Dose giorn. di l-D –10-30%] preparazioni a rilascio lento, transdermiche, sottocutanee, nanoparticles lipidiche solide e microparticles biocompatibili. infusione continua digiunale.")
37
è corretta l’idea classica che non si deve iniziare da subito con l-DOPA, perché il trattamento perde efficacia dopo qualche anno? NO il trattamento perde efficacia perché prosegue la perdita neuronale i disturbi motori (ON/OFF, discinesie/acinesia) sono dovuti a perdita di capacità di accumulare dopamina e rilasciare nel tempo non si usano più anticolinergici / amantadina (casomai agonisti colinergici nicotinici) uso iniziale di agonisti invece di l-DOPA: sembra mostrare minore performance motoria ma qualche complicazione motoria in meno – si associa invece a maggior sonnolenza (trial con pramipexolo) uso di agonisti insieme a l-DOPA: vedi sopra riduzione delle fluttuazioni ON-OFF
 sono dovuti a perdita di capacità di accumulare dopamina e rilasciare nel tempo. non si usano più anticolinergici / amantadina (casomai agonisti colinergici nicotinici) uso iniziale di agonisti invece di l-DOPA: sembra mostrare minore performance motoria ma qualche complicazione motoria in meno – si associa invece a maggior sonnolenza (trial con pramipexolo) uso di agonisti insieme a l-DOPA: vedi sopra riduzione delle fluttuazioni ON-OFF.")
38
fino a pochi anni fa: la l-DOPA può essere tossica agonisti DAergici possono avere effetti neuroprotettivi in assenza di demenza/psicosi vanno usati come trattamento iniziale farmaci DAergici ora: la l-DOPA non accelera la progressione della malattia non ci sono vantaggi dell’uno o altro trattamento in termini di neuroprotezione la l-DOPA è più efficace degli agonisti sui sintomi motori e nel migliorare le attività quotidiane DA-agonisti associati a meno complicazioni motorie DA-agonisti associati a più effetti sgraditi (allucinazioni e sonnolenza)meno complicazioni motorie Nessuna evidenza di benefici a lungo termine con terapia iniziale con DA-agonisti
meno complicazioni motorie. Nessuna evidenza di benefici a lungo termine con terapia iniziale con DA-agonisti.")
39
altri approcci: agonisti colinergici l-DOPA allevia efficacemente sintomi motori ma non agisce efficacemente su deficit cognitivi agenti nicotinici appaiono essere attivi in questo senso – nicotina e SIB-1553A migliorano performance in test cognitivi e se co-sommnistrati contrastano i deficit indotti da l-DOPA altri target farmacologici e terapie neuroprotettive – glial cell line-derived neurotrophic factor (GDNF) – neuroimmunofiline – agenti attivi sullo stress ossidativo: minociclina, Coenzima Q10, creatina, glutatione ridotto – antagonisti del recettore A2A per l’adenosina – inibitori del rilascio di glutammato (eccitotossicità?) – approcci antinfiammatori – trapianti e terapie geniche, stem cells (regimi dietetici, fitofarmaci, antiossidanti) ??
 – neuroimmunofiline. – agenti attivi sullo stress ossidativo: minociclina, Coenzima Q10, creatina, glutatione ridotto. – antagonisti del recettore A2A per l’adenosina. – inibitori del rilascio di glutammato (eccitotossicità ) – approcci antinfiammatori. – trapianti e terapie geniche, stem cells. (regimi dietetici, fitofarmaci, antiossidanti)")
40
target promettenti nuclear factor erythroid-2-related factor 2 (Nrf2): fattore di trascrizione che induce espressione di enzimi ad azione citoprotettiva, antixenobiotica e antiossidante (eme-ossigenasi-1, NAD(P)H:chinone ossidoreduttasi) e enzimi del metabolismo del glutathione (GSH) (gamma-glutamil cisteina ligasi, GSH transferasi) due strategie per aumentare attività trascrizionale di Nrf2: – chinoni derivati dal catecolo per inibire selettivamente la proteina Kelch-like associata a ECH, repressore di Nrf2 (per aumentare i livelli della proteina) – inibitori della glicogeno sintetasi chinasi 3beta (per alzare i livelli di proteina e di attività nel nucleo)
: fattore di trascrizione che induce espressione di enzimi ad azione citoprotettiva, antixenobiotica e antiossidante (eme-ossigenasi-1, NAD(P)H:chinone ossidoreduttasi) e enzimi del metabolismo del glutathione (GSH) (gamma-glutamil cisteina ligasi, GSH transferasi) due strategie per aumentare attività trascrizionale di Nrf2: – chinoni derivati dal catecolo per inibire selettivamente la proteina Kelch-like associata a ECH, repressore di Nrf2 (per aumentare i livelli della proteina) – inibitori della glicogeno sintetasi chinasi 3beta (per alzare i livelli di proteina e di attività nel nucleo)")
41
target promettenti Glicoproteina-P (P-g, trasportatore ABC-B1): trasportatore della barriera E.E. che limita accumulo di xenobiotici nel cervello Infiammazione e infezione alterano i livelli di espressione e attività di P-g (down-regulation da mediatori infiammatori a breve termine, up-regulation a lungo termine) sistema cannabico – possibile ruolo protettivo del sistema degli endocannabinoidi – ruolo citoprotettivo del cannabidiolo (composto naturale privo di azione psicotropa) perché agonista potrebbe in teoria essere “neuroprotettivo”?
: trasportatore della barriera E.E. che limita accumulo di xenobiotici nel cervello Infiammazione e infezione alterano i livelli di espressione e attività di P-g (down-regulation da mediatori infiammatori a breve termine, up-regulation a lungo termine) sistema cannabico – possibile ruolo protettivo del sistema degli endocannabinoidi – ruolo citoprotettivo del cannabidiolo (composto naturale privo di azione psicotropa) perché agonista potrebbe in teoria essere neuroprotettivo")
42
target promettenti GLIA la malattia di Parkinson può progredire anche quando viene a mancare la causa iniziale di neurodegenerazione. Sostanze tossiche di origine gliale possono contribuire a perpetuare il quadro degenerativo: – citochine proinfiammatorie (TNF-alpha, Il-1beta, IFN-gamma) possono stimulare produzione gliale di NO o esercitare effetti diretti sui neuroni dopaminergici attivando recettori con domini “death” intracitoplasmici e => apoptosi inibizione di caspasi o recettori di TNF-alpha non funzionano nei modelli sperimentali, suggerendo che non basta interrompere un solo percorso di trasduzione pioglitazone (agonista PPAR-gamma [*]) e minociclina (derivato tetraciclinico) riducono attivazione gliale e proteggono SN in modelli animali [* peroxisome proliferator-activated receptor-gamma]
 possono stimulare produzione gliale di NO o esercitare effetti diretti sui neuroni dopaminergici attivando recettori con domini death intracitoplasmici e => apoptosi. inibizione di caspasi o recettori di TNF-alpha non funzionano nei modelli sperimentali, suggerendo che non basta interrompere un solo percorso di trasduzione pioglitazone (agonista PPAR-gamma [*]) e minociclina (derivato tetraciclinico) riducono attivazione gliale e proteggono SN in modelli animali. [* peroxisome proliferator-activated receptor-gamma]")
43
DEEP BRAIN STIMULATION
impianto di elettrodi permanenti in STN stimolazione ad alta frequenza con effetto di silenziamento del STN: riequilibrio dei circuiti interni, sbilanciati verso l’inibizione (via “indiretta”) con drammatico miglioramento clinico possibili effetti imprevisti per posizionamento impreciso o stimolazione imprevista di regioni e strutture vicine
 con drammatico miglioramento clinico. possibili effetti imprevisti per posizionamento impreciso o stimolazione imprevista di regioni e strutture vicine.")
44
i problemi a lungo termine
Discinesie: diversi lavori suggeriscono che in trattamento con l-DOPA a lungo termine, i terminali serotoninergici striatali possano rilasciare DA e possano costituire il principale determinante presinaptico delle discinesie da l-DOPA sistema serotoninico come target di terapia anti-discinetica ? Dopamine dysregulation syndrome (DDS): disturbo iatrogeno a lungo termine: ricorso compulsivo nel 3-4% dei casi a terapia dopaminergica sostitutiva (DRT). Disturbo dei meccanismi decisionali, influenzati da farmaci dopaminergici, favorito dalla compromissione dei collegamenti crociati tra striato e corteccia frontale. Gambling (gioco d’azzardo) patologico (o anche shopping patologico ecc.) – anch’esso legato a trattamento (farmacologico o deep-brain stimulation), e presumibilmente ad alterazione dei processi gratificazionali DA-dipendenti
: disturbo iatrogeno a lungo termine: ricorso compulsivo nel 3-4% dei casi a terapia dopaminergica sostitutiva (DRT). Disturbo dei meccanismi decisionali, influenzati da farmaci dopaminergici, favorito dalla compromissione dei collegamenti crociati tra striato e corteccia frontale. Gambling (gioco d’azzardo) patologico (o anche shopping patologico ecc.) – anch’esso legato a trattamento (farmacologico o deep-brain stimulation), e presumibilmente ad alterazione dei processi gratificazionali DA-dipendenti.")
45
NOTA su altri farmaci vi sono molti farmaci che possono esacerbare lo stress ossidativo: è cruciale ricordare che possono determinare declino cognitivo irreversibile e/o Parkinson. tra questi farmaci si ricorda l’antiepilettico valproato che presumibilmente interferisce con il funzionamento del complesso respiratorio I. in presenza di segni di compromissione cognitiva o parkinsonismo tali farmaci vanno ovviamente sospesi.








 STEATOSI CIRROSI (8-25%) HCC.>")









![L-DOPA [(-)- o l-DOPA] Terapia sostitutiva (indiretta)](/2/579607/big_thumb.jpg "L-DOPA [(-)- o l-DOPA] Terapia sostitutiva (indiretta)>")


