Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Cromatografia

2
Cromatografia Il termine cromatografia indica un insieme di tecniche che hanno lo scopo di separare una miscela nei suoi componenti, per permetterne il riconoscimento qualitativo e quantitativo Queste tecniche sono basate sulla distribuzione differenziale dei vari componenti fra due fasi, una chiamata fase fissa o fase stazionaria e l’altra chiamata fase mobile o eluente, che fluisce in continuo attraverso la fase fissa Le tecniche sono molto utilizzate in campo archeometrico, essendo particolarmente utili nell’analisi di miscele complesse come sono la maggior parte dei campioni di natura organica

3
Nascita della cromatografia
inizi del XX secolo come tecnica per la separazione di pigmenti fogliari, inventata dal botanico russo Mikhail Semenovich Tswett. Egli intendeva separare i pigmenti presenti nella clorofilla; fece un estratto di foglie verdi in etere di petrolio, lo depositò in testa ad una colonna di vetro impaccata con carbonato di calcio ed eluì, (cioè versò in continuo) con solfuro di carbonio: i vari pigmenti si separano in bande colorate, in particolare clorofilla A e B, carotene e xantofilla Tswett chiamò questa tecnica cromatografia dal greco scrittura del colore
 con solfuro di carbonio: i vari pigmenti si separano in bande colorate, in particolare clorofilla A e B, carotene e xantofilla. Tswett chiamò questa tecnica cromatografia dal greco scrittura del colore.")
4
Cenni preliminari Le tecniche cromatografiche sono sempre distruttive (anche se in senso strettamente analitico possono in alcuni casi essere non distruttive), in quanto operano esclusivamente su campioni in soluzione o in fase vapore: i materiali oggetto di analisi vanno quindi disciolti in un opportuno solvente. Non è possibile l’analisi senza prelievo di campione né tanto meno l’analisi in situ (tranne con strumenti miniaturizzati) è bene precisare che il consumo di campione è minimo. Sono sufficienti da 1 ml a 1 µl di soluzione, corrispondenti a pochi mg di campione solido
, in quanto operano esclusivamente su campioni in soluzione o in fase vapore: i materiali oggetto di analisi vanno quindi disciolti in un opportuno solvente. Non è possibile l’analisi senza prelievo di campione né tanto meno l’analisi in situ (tranne con strumenti miniaturizzati) è bene precisare che il consumo di campione è minimo. Sono sufficienti da 1 ml a 1 µl di soluzione, corrispondenti a pochi mg di campione solido.")
5
Visualizzazione della separazione
Ponendo all’uscita della colonna un rivelatore che misuri la concentrazione del soluto nell’eluito (cioè la fase mobile che esce dalla colonna) e riportando il segnale in funzione del tempo si può ottenere un cromatogramma La posizione dei picchi sull’asse dei tempi, o tempo di ritenzione, serve per identificare i componenti del campione L’area sottesa dai picchi è proporzionale alla quantità di ogni singolo componente e può essere utilizzata a scopo quantitativo
 e riportando il segnale in funzione del tempo si può ottenere un cromatogramma. La posizione dei picchi sull’asse dei tempi, o tempo di ritenzione, serve per identificare i componenti del campione. L’area sottesa dai picchi è proporzionale alla quantità di ogni singolo componente e può essere utilizzata a scopo quantitativo.")
6
Tempo di ritenzione Il tempo di ritenzione tR è il tempo che impiega un componente della miscela iniettata ad uscire dalla colonna o, tecnicamente, ad essere rivelato come picco dal detector. Un tipico cromatogramma per una miscela a due componenti ha due situazioni diverse: il picco a sinistra rappresenta un soluto che non ha alcuna interazione con la fase stazionaria ed esce al cosiddetto tempo morto, tM il picco a destra rappresenta un soluto che ha, invece, interazione con la fase stazionaria ed esce al tempo tR > tM

7
Tempo di ritenzione Oltre al tempo di ritenzione tR, è possibile quantificare l’interazione di un soluto con la fase stazionaria in due modi: mediante il volume di fase mobile VR necessario per eluire il soluto, VR = tR x F ; F = velocità di flusso mediante il fattore di capacità K’, espresso come la differenza tra il tempo di ritenzione ed il tempo morto in unità di tempo morto:

8
Piatti teorici Il sistema cromatografico è immaginato come una colonna composta da una serie di strati sottili chiamati piatti teorici; in ognuno di questi microelementi della colonna si realizza l’equilibrio di distribuzione del soluto tra fase stazionaria e fase mobile. Lo spostamento del soluto lungo la colonna è dovuto all’azione dinamica della fase mobile I termini numero di piatti teorici (N) e altezza del piatto (HETP, Height Equivalent to Theoric Plate) sono comunemente utilizzati in cromatografia per quantificare le prestazioni dei sistemi cromatografici HETP = lunghezza colonna /N
 e altezza del piatto (HETP, Height Equivalent to Theoric Plate) sono comunemente utilizzati in cromatografia per quantificare le prestazioni dei sistemi cromatografici. HETP = lunghezza colonna /N.")
9
Separazione ottimale separazione con scarsa risoluzione e basso N migliora la risoluzione ma è sempre basso N ottima risoluzione e buono N

10
Interazione soluto-fasi
Le interazioni che si verificano tra le sostanze da separare e le due fasi (mobile e stazionaria) sono deboli: se così non fosse non ci sarebbe trattenimento sulla fase stazionaria oppure, al contrario, eluizione. Sono sfruttate a scopo separativo le seguenti interazioni: In tutte queste interazioni svolge un ruolo solitamente decisivo la polarità delle due fasi. Spesso possono essere presenti più tipi di interazione nello stesso processo cromatografico legami a idrogeno interazioni dipolo-dipolo interazioni dipolo-dipolo indotto forze di Van der Waals formazione di composti di interazione attrazione coulombiana interazioni steriche
 sono deboli: se così non fosse non ci sarebbe trattenimento sulla fase stazionaria oppure, al contrario, eluizione. Sono sfruttate a scopo separativo le seguenti interazioni: In tutte queste interazioni svolge un ruolo solitamente decisivo la polarità delle due fasi. Spesso possono essere presenti più tipi di interazione nello stesso processo cromatografico. legami a idrogeno. interazioni dipolo-dipolo. interazioni dipolo-dipolo indotto. forze di Van der Waals. formazione di composti di interazione. attrazione coulombiana. interazioni steriche.")
11
Meccanismi della separazione
In base ai tipi di interazione prima descritti possiamo suddividere i meccanismi di separazione impiegati in cromatografia in: adsorbimento ripartizione scambio ionico esclusione affinità

12
Adsorbimento La fase stazionaria è un solido in polvere steso su un supporto; sulla superficie dei granuli si trovano siti attivi che possono stabilire legami deboli (reversibili!) con le molecole della miscela da separare. Si parla quindi di cromatografia di adsorbimento, che può essere gas-solido o liquido-solido a seconda della natura della fase mobile La cromatografia di adsorbimento è utilizzata per separare sostanze neutre polari o non polari, di natura organica o inorganica
 con le molecole della miscela da separare. Si parla quindi di cromatografia di adsorbimento, che può essere gas-solido o liquido-solido a seconda della natura della fase mobile. La cromatografia di adsorbimento è utilizzata per separare sostanze neutre polari o non polari, di natura organica o inorganica.")
13
Ripartizione La fase stazionaria è un liquido che impregna un solido granulare inerte o è ad esso chimicamente legato; in questo liquido le molecole da separare sono solubili; la fase stazionaria e la fase mobile devono invece essere immiscibili. Durante l’eluizione le molecole si ripartiscono dinamicamente tra le due fasi secondo la diversa solubilità di ognuna. Si parla quindi di cromatografia di ripartizione, che può essere gas-liquido o liquido-liquido a seconda della natura della fase mobile La cromatografia di ripartizione è chiamata in fase normale se la fase stazionaria è più polare della fase mobile, mentre è chiamata fase inversa se la fase stazionaria è meno polare della fase mobile. Si tratta della tecnica più comunemente impiegata per la separazione di sostanze organiche

14
Scambio ionico La fase stazionaria è costituita da un polimero inerte contenente siti attivi ionizzati o ionizzabili, i cui controioni possono essere scambiati con altri ioni aventi carica dello stesso segno. Il meccanismo di separazione è basato sulla competizione per i siti di scambio tra gli ioni presenti nella fase mobile e quelli presenti nel campione. Si parla di cromatografia di scambio ionico (IEC) La cromatografia a scambio ionico è impiegata per la separazione di sostanze ioniche o ionizzabili
 La cromatografia a scambio ionico è impiegata per la separazione di sostanze ioniche o ionizzabili.")
15
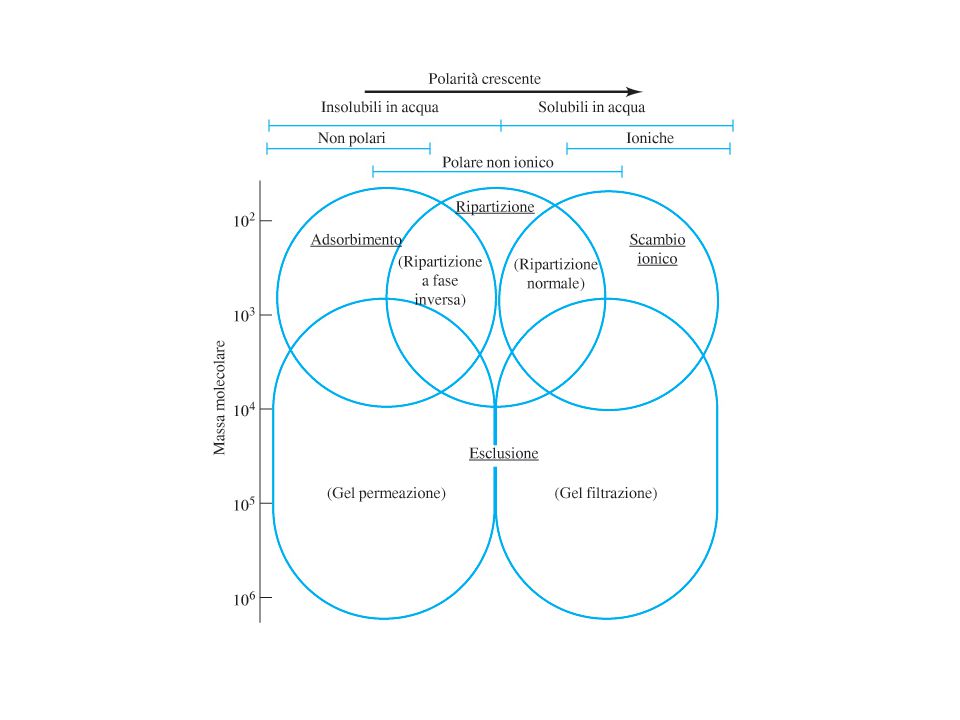
Esclusione dimensionale
La fase stazionaria è un solido poroso o un gel. Le molecole dell’analita, disciolte nella fase mobile, penetrano nei pori se le loro dimensioni sono compatibili e vi rimangono per un certo tempo; le molecole più grandi sono invece escluse dai pori ed escono dalla colonna in tempi brevi Si parla di cromatografia di esclusione dimensionale (SEC) Gel permeazione per la separazione di sostanze insolubili in acqua Gel filtrazione per la separazione di sostanze solubili in acqua La tecnica è impiegata per la separazione di molecole di grandi dimensioni
 Gel permeazione per la separazione di sostanze insolubili in acqua. Gel filtrazione per la separazione di sostanze solubili in acqua. La tecnica è impiegata per la separazione di molecole di grandi dimensioni.")
16
Affinità In questo caso si utilizzano reazioni di tipo biochimico, reversibili e molto specifiche, in modo che le molecole da separare interagiscano con la fase stazionaria e si ottenga così l’eluizione selettiva di alcuni componenti della miscela. Si parla di cromatografia di affinità (AFC) La cromatografia di affinità è impiegata nella separazione di molecole di interesse prevalentemente biochimico
 La cromatografia di affinità è impiegata nella separazione di molecole di interesse prevalentemente biochimico.")
18
Stato fisico della fase mobile
In base allo stato fisico della mobile possiamo classificare le tecniche cromatografiche come segue: Cromatografia Liquida (LC): la fase mobile è un liquido nel quale siano solubili i componenti della miscela da separare; la fase stazionaria deve essere insolubile nella fase mobile Gascromatografia (GC): la fase mobile è un gas che funge da carrier per i componenti della miscela Cromatografia fluida supercritica (SFC): la fase mobile è un fluido supercritico, con proprietà intermedie tra un liquido e un gas
: la fase mobile è un liquido nel quale siano solubili i componenti della miscela da separare; la fase stazionaria deve essere insolubile nella fase mobile. Gascromatografia (GC): la fase mobile è un gas che funge da carrier per i componenti della miscela. Cromatografia fluida supercritica (SFC): la fase mobile è un fluido supercritico, con proprietà intermedie tra un liquido e un gas.")
19
Forma del letto cromatografico
In base alla forma del letto cromatografico su cui è realizzato il processo separativo, possiamo le seguenti varianti: Cromatografia su colonna: la fase stazionaria è contenuta all’interno di una colonna cilindrica, che può riempire completamente (colonna impaccata) oppure rivestirne la superficie interna (colonna tubulare) Cromatografia planare: la fase stazionaria è distribuita su una superficie piana, che può essere un supporto cartaceo (cromatografia su carta, PC) o una lastrina in vetro o altri materiali (cromatografia su strato sottile, TLC)
 oppure rivestirne la superficie interna (colonna tubulare) Cromatografia planare: la fase stazionaria è distribuita su una superficie piana, che può essere un supporto cartaceo (cromatografia su carta, PC) o una lastrina in vetro o altri materiali (cromatografia su strato sottile, TLC)")
20
Tecniche cromatografiche
In base alla forma del letto cromatografico Cromatografia su colonna (impaccata, open-tubular) Cromatografia planare (su carta, su strato sottile) In base allo stato fisico della fase mobile Cromatografia Liquida (LC) Gascromatografia (GC) Cromatografia fluida supercritica (SFC) In base al meccanismo di separazione Adsorbimento Ripartizione Scambio ionico Esclusione Affinità
 Cromatografia planare (su carta, su strato sottile) In base allo stato fisico della fase mobile. Cromatografia Liquida (LC) Gascromatografia (GC) Cromatografia fluida supercritica (SFC) In base al meccanismo di separazione. Adsorbimento. Ripartizione. Scambio ionico. Esclusione. Affinità.")
21
Cromatografia liquida
La cromatografia liquida è impiegata per la separazione di sostanze non volatili, neutre o ioniche, e di sostanze termolabili. Si presta facilmente a misure quantitative. Si possono separare sostanze appartenenti a varie classi tra cui, di interesse archeometrico: aminoacidi, peptidi e proteine idrocarburi carboidrati terpenoidi ioni inorganici Cromatografia planare Si tratta di un gruppo di tecniche di cromatografia liquida di semplicissima applicazione, spesso impiegate per avere informazioni preliminari. La fase stazionaria è supportata su lastre di vetro, fogli di alluminio o di plastica nella versione TLC (Thin Layer Chromatography) e su fogli di carta da filtro nella versione PC (Paper Chromatography) Le fasi stazionarie più usate sono il gel di silice e l’allumina per la cromatografia di adsorbimento, la cellulosa per la ripartizione liquido-liquido (in questo caso la fase stazionaria è l’acqua adsorbita sulle particelle di cellulosa)
 e su fogli di carta da filtro nella versione PC (Paper Chromatography) Le fasi stazionarie più usate sono il gel di silice e l’allumina per la cromatografia di adsorbimento, la cellulosa per la ripartizione liquido-liquido (in questo caso la fase stazionaria è l’acqua adsorbita sulle particelle di cellulosa)")
22
Cromatografia planare
L’esecuzione dell’analisi è molto semplice: la miscela da separare va depositata sulla superficie, posandone con un tubo capillare una goccia su una linea che segna l’inizio del processo di eluizione Quindi il foglio o la lastrina si pongono in una vaschetta contenente la fase mobile che per gravità (modalità discendente), per capillarità (modalità ascendente) o per diffusione laterale (modalità orizzontale) fluisce sulla fase fissa trascinando gli analiti e separandoli
, per capillarità (modalità ascendente) o per diffusione laterale (modalità orizzontale) fluisce sulla fase fissa trascinando gli analiti e separandoli.")
23
Cromatografia planare
(a) camera di sviluppo a flusso ascendente (b) camera di sviluppo a flusso orizzontale
 camera di sviluppo a flusso ascendente. (b) camera di sviluppo a flusso orizzontale.")
24
Cromatografia planare
Il risultato è (spesso ma non sempre) visualizzabile sotto forma di macchie colorate, ognuna dovuta ad un componente della miscela. Il riconoscimento delle sostanze può avvenire effettuando separazioni su miscele standard; in questo caso il parametro che caratterizza i soluti separati è il cosiddetto Rf o fattore di ritardo. Per ogni analita il valore di Rf si ottiene misurando la distanza percorsa dal centro della macchia e confrontandola con la distanza percorsa dal fronte dell’eluente: Rf = danalita / deluente Il valore di Rf degli analiti è quindi sempre compreso tra 0 e 1. I valori ottimali sono compresi tra 0.4 e 0.8
 visualizzabile sotto forma di macchie colorate, ognuna dovuta ad un componente della miscela. Il riconoscimento delle sostanze può avvenire effettuando separazioni su miscele standard; in questo caso il parametro che caratterizza i soluti separati è il cosiddetto Rf o fattore di ritardo. Per ogni analita il valore di Rf si ottiene misurando la distanza percorsa dal centro della macchia e confrontandola con la distanza percorsa dal fronte dell’eluente: Rf = danalita / deluente. Il valore di Rf degli analiti è quindi sempre compreso tra 0 e 1. I valori ottimali sono compresi tra 0.4 e 0.8.")
25
TLC Quando non si hanno informazioni preliminari circa la composizione della matrice, l’utilizzo della TLC su piccole quantità fornisce un utile aiuto per l’elaborazione della migliore strategia di separazione ed identificazione, poiché il comportamento può essere replicato abbastanza fedelmente impiegando una fase mobile ed una stazionaria simili in un procedimento di cromatografia su colonna umida o a secco.

26
Nella TLC si impiegano lastre di supporto (vetro, alluminio, poliestere) rivestite di gel di silice o allumina o polvere di cellulosa contenenti in molte di esse un composto fluorescente che serve, una volta separati i componenti della miscela, a rivelarli più facilmente quando la lastra viene esposta alla luce ultravioletta. Il campione viene deposto con un capillare in vetro in modo che non venga caricata una quantità di campione superiore agli 0.5 l così da generare una macchia di circa 3mm di diametro. Si lascia quindi eluire il campione sotto l’azione del solvente che sale, in una cameretta satura di vapori dello stesso,per capillarità. Quando sono stati raggiunti i 4/5 della lastra si tratta il cromatogramma con un opportuno rivelatore delle sostanze da separare e dosare (irradiamento con UV, vapori di iodio, carbonizzazione con ac. Solforico, reagenti cromogeni specifici per gruppi funzionali ).
..")
27
Nel caso della TLC Rf = distanza percorsa dalla sostanza /distanza percorsa dal solvente tuttavia una reale riproducibilità di Rf risulta difficile da ottenere a causa di piccole variazioni di fattori quali:la grandezza delle particelle dell’adsorbente, la composizione del solvente ed il grado di saturazione nella vaschetta di eluizione, spessore delle lastre ecc. Per tutti i motivi su citati , dunque, non è opportuno utilizzare i valori di Rf come criteri identificativi ma sempre che siano disponibili, porre sulle lastre anche degli standard a cui far riferimento. Da un punto di vista quantitativo la valutazione può essere effettuata o valutando le proprietà fisiche correlandole alla concentrazione o misurare dopo dissoluzione della parte del cromatogramma in questione in un adatto solvente.

28
Cromatografia su carta

29
Visualizzazione dei risultati
Nel caso le macchie non siano colorate, è possibile ricorrere a due metodi per visualizzare il risultato della separazione: utilizzare una lampada UV per irraggiare la lastrina, se le sostanze separate non assorbono la luce visibile ma assorbono nell’ultravioletto (l < 400 nm); può essere necessario addizionare alla fase stazionaria o alla fase mobile un indicatore di fluorescenza che permette di localizzare le macchie spruzzare la lastrina con una soluzione contenente sostanze in grado di reagire con i costituenti della miscela separata, generando composti colorati; può essere necessario scaldare leggermente la lastrina per favorire la reazione
; può essere necessario addizionare alla fase stazionaria o alla fase mobile un indicatore di fluorescenza che permette di localizzare le macchie. spruzzare la lastrina con una soluzione contenente sostanze in grado di reagire con i costituenti della miscela separata, generando composti colorati; può essere necessario scaldare leggermente la lastrina per favorire la reazione.")
30
Irraggiamento con UV Separazione di coloranti antrachinonici con TLC e illuminazione con lampada UV a 254 nm (dx); l’intensità delle macchie può essere valutata con un colorimetro (sotto)
; l’intensità delle macchie può essere valutata con un colorimetro (sotto)")
31
Addizione di reagenti cromogeni
Nell’immagine sotto è mostrato un contenitore per l’aspersione di ninidrina su lastrine TLC o PC Alcuni esempi di reagenti correntemente impiegati per evidenziare le macchie sono riportati nella tabella sottostante Reagente Utilizzo Iodio in EtOH per composti azotati AgNO3 in NH3 per sostanze riducenti Alizarina per cationi Ninidrina per amminoacidi e ammine HS2 per cationi e metalli pesanti

32
Cromatografia planare bidimensionale
Per aumentare la separazione tra gli analiti e quindi la loro identificazione è possibile effettuare l’eluizione lungo due dimensioni, prima lungo un asse e poi, girando a 90° la lastrina, lungo l’asse ortogonale, evenutalmente con una fase mobile differente: in questo modo le macchie sono separate in maniera più efficiente Sotto: separazione di aminoacidi

33
Esempio: pigmenti fogliari
Nella figura è mostrata la separazione per cromatografia su carta orizzontale di pigmenti fogliari da un estratto della pianta Cisto bianco; si tratta dei pigmenti che fanno cambiare il colore delle foglie nelle diverse stagioni. La separazione è ottenuta con un disco di carta da filtro come fase fissa e alcol etilico al 95 % come fase mobile. Gli aloni sono attribuiti alle diverse sostanze sulla base del loro comportamento chimico: le sostanze più idrosolubili (e quindi più affini all’acqua adsorbita sulla cellulosa, che costituisce la fase fissa) sono quelle al centro, cioè clorofille A e B; le sostanze meno idrosolubili, xantofilla e carotina, migrano all’esterno in quanto più affini alla fase mobile
 sono quelle al centro, cioè clorofille A e B; le sostanze meno idrosolubili, xantofilla e carotina, migrano all’esterno in quanto più affini alla fase mobile.")
34
Separazione di coloranti porpora
Separazione per PC di estratti in etanolo da molluschi I molluschi sono delle specie Dicathais orbita, Murex brandaris, Murex trunculus, Purpura haemastoma, Murex erinaceus e Rapana bezoar, tutti impiegati in antichità per ottenere coloranti porpora. I valori di Rf sono confrontati con un composto indigoide, il potassio indossil solfato (K.I.S., a sinistra)
")
35
Cromatografia liquida su colonna
I primi esperimento di cromatografia su colonna utilizzavano colonne di vetro di 1-5 cm di diametro e lunghezza fino a 5 metri. Ciò richiedeva tempi diseparazione molto lunghi Attualmente è possibile realizzare microcolonne di pochi cm di lunghezza, in grado di separare in pochi minuti molte sostanze. Queste colonne sono costituite da particelle di 1-5 µm di diametro, che richiedono pressioni molto alte per forzare il passaggio della fase mobile attraverso la colonna. Per sistemi di questo genere il termine utilizzato è cromatografia liquida ad elevate prestazioni o elevate pressioni (HPLC, High Performance o Pressure Liquid Chromatography)
")
36
Metodi di ripartizione
[S]A = 0.09 [S]B = 0.91 [S]A = 1

37
EQUILIBRIO FASE MOBILE A FASE STAZIONARIA A

38
EQUILIBRIO FASE MOBILE A FASE STAZIONARIA A

39
EQUILIBRIO FASE MOBILE S S FASE STAZIONARIA S S

40
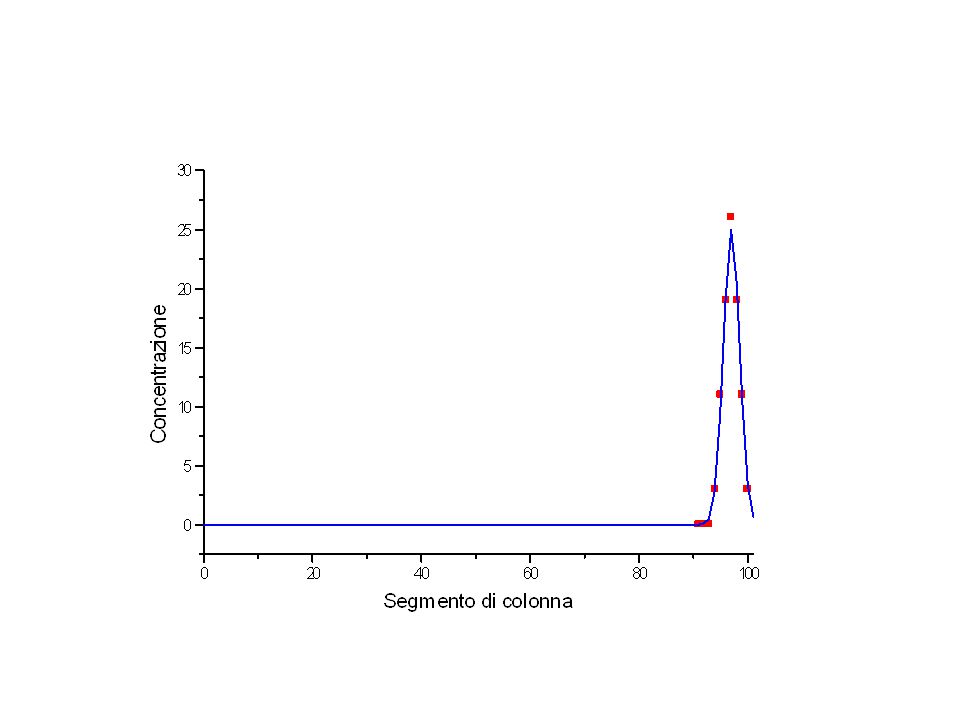
50 50 25 12.5 37.5 6 38 25 100
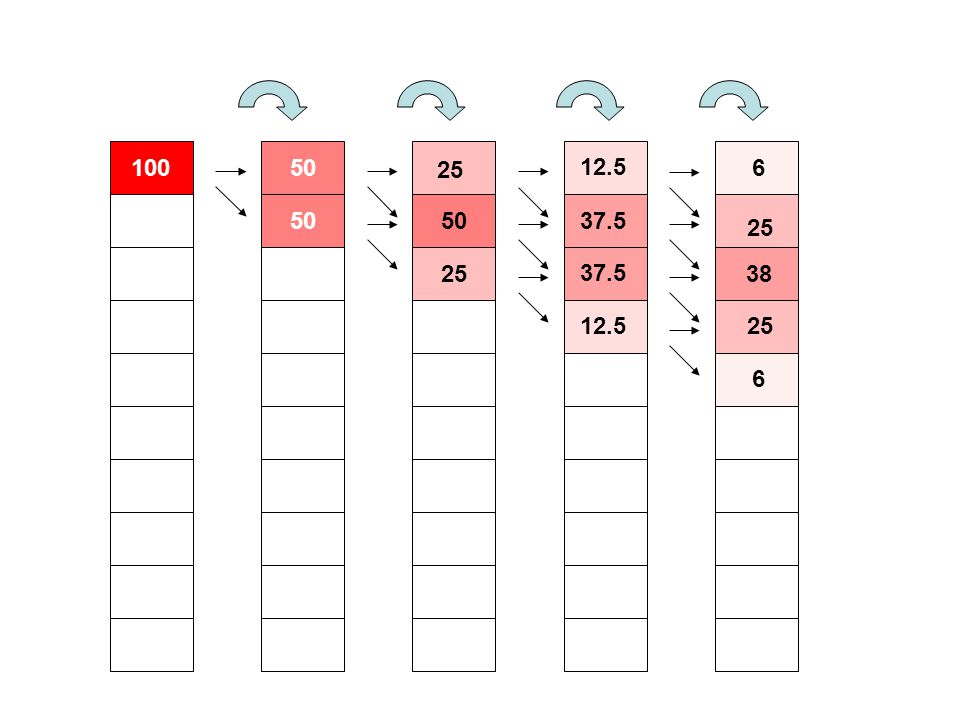
41
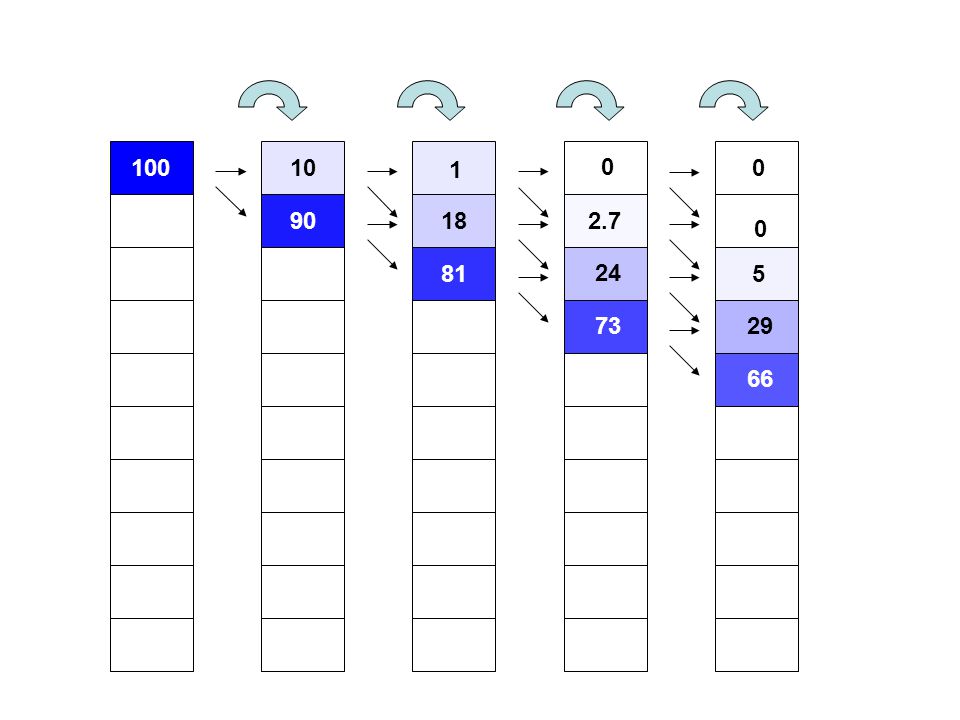
100 10 1 90 18 2.7 81 24 5 73 29 66
42
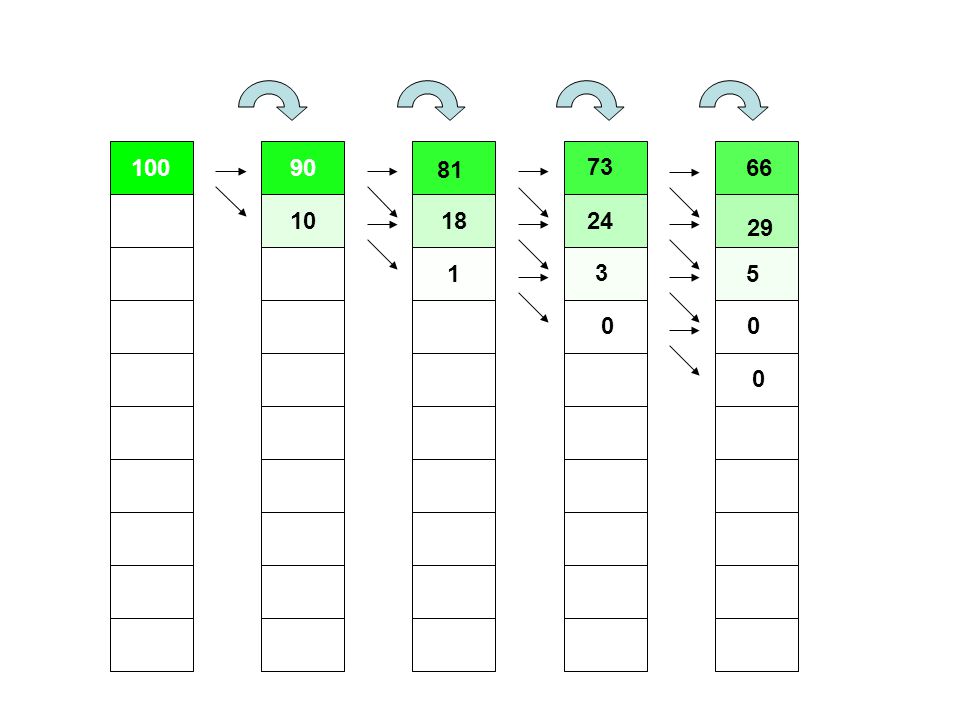
100 90 81 73 66 10 18 24 29 1 3 5
43
100

46
Cromatografia su colonna

47
Cromatografia liquida su colonna
I primi esperimento di cromatografia su colonna utilizzavano colonne di vetro di 1-5 cm di diametro e lunghezza fino a 5 metri. Ciò richiedeva tempi diseparazione molto lunghi Attualmente è possibile realizzare microcolonne di pochi cm di lunghezza, in grado di separare in pochi minuti molte sostanze. Queste colonne sono costituite da particelle di 1-5 µm di diametro, che richiedono pressioni molto alte per forzare il passaggio della fase mobile attraverso la colonna. Per sistemi di questo genere il termine utilizzato è cromatografia liquida ad elevate prestazioni o elevate pressioni (HPLC, High Performance o Pressure Liquid Chromatography)
")
48
Rapporto di ripartizione
Volume di ritenzione Rapporto di ripartizione β

49
Per una specie non ritenuta: VR = VM e k’ = 1

50
Risoluzione A metà altezza: wh = w

51
Risoluzione

52
Piatti teorici e larghezza dei picchi, capacità di picchi

53
Adsorbimento (L.S.C.) Questa tecnica è legata all'adsorbimento di un soluto su un adsorbente polare come: gel di silice, allumina, farina fossile, ossido di magnesio e carbone o altri composti che abbiano le stesse caratteristiche fisiche. Il meccanismo che regola questo tipo di separazione è da attribuirsi alla competizione dei soluto tra i gruppi polari presenti sulla superficie della fase stazionaria e la fase mobile non polare.

54
Adsorbimento (L.S.C.) Fra le tecniche di separazione è stata per lungo tempo la più usata per l'alta risoluzione nella separazione con l'impiego di solventi apolari. Con l'introduzione della fase inversa è stato possibile migliorare questa tecnica impiegando nella fase di eluizione dei solventi polari (acquosi) al contrario della fase diretta dove l'acqua ha un'azione disattivante sulla fase stazionaria.
 al contrario della fase diretta dove l acqua ha un azione disattivante sulla fase stazionaria.")
55
Adsorbimento

56
Fase normale (NP) Fase stazionaria polare (es., silica, alumina)
Fase mobile non polare(es., esano, cloruro di metilene) Separa composti polari
 Separa composti polari.")
57
Fase normale (NP) Il materiale adsorbente più comunemente usato è il gel di silice che è preparato facendo reagire silicato di sodio con un acido minerale, Ne risulta una polimerizzazione con formazione di un sistema tridimensionale di tetraedri di Si04

58
Fase inversa (RP) Fase stazionaria non polare (e.g., C18, C8)
Fase mobile polare (e.g., acqua, MeOH, Separa composti non polari

59
Fasi legate Fasi stazionaria legate chimicamente ad un supporto inerte per produrre fasi idroliticamente stabili

60
Fasi legate Cromatografia a fase normale
Il letto stazionario è polare, la fase mobile è non polare o meno polare Cromatografia a fase inversa Il letto stazionario è apolare, la fase mobile è polare o più polare

61
Esclusione (SEC) 2 tipi : − Gel filtration − Gel permeation
Fase stazionaria è silica o un polimero con dimensione dei pori controllata Fase mobile semplicemente scioglie e trasporta il campione attraverso il sistema

62
Tipi di matrici utilizzate
Agarosio Polisaccaride derivante da particolari alghe rosse cosituito da residui alternati di D-galattoso e 3,6-anidro-L-galattoso. Disponibile commercialmente come Sepharose, Sepharose CL (cross-linking con 2,3-dibromopropanolo), Superose, Bio-Gel A. Limiti di esclusione kD. Cellulosa Polimero di unità glucosidiche unite da legami -1,4. Tramite epicloridrina vengono ottenuti i legami crociati essenziali per l’utilizzo come matrice in cromatografia, il numero dei quali determina la dimensione dei pori. Destrano Polimero di residui di glucosio uniti da legami -1,6 prodotto dal batterio Leuconostoc mesenteroides. In commercio con il nome di Sephadex. La porosità dei gel a base di destrano è controllata dalla massa molecolare del destrano usato e dall’ introduzione di legami crociati ottenuti con epicloridrina. Limiti di esclusione kD Il Sephacryl è destrano con legami crociati ottenuti con N,N’-metilene bisacrilamide. Limiti di esclusione kD. Il Superdex e’ un gel composito costituito da destrano covalentemente legato ad agarosio.
, Superose, Bio-Gel A. Limiti di esclusione kD. Cellulosa Polimero di unità glucosidiche unite da legami -1,4. Tramite epicloridrina vengono ottenuti i legami crociati essenziali per l’utilizzo come matrice in cromatografia, il numero dei quali determina la dimensione dei pori. Destrano Polimero di residui di glucosio uniti da legami -1,6 prodotto dal batterio Leuconostoc mesenteroides. In commercio con il nome di Sephadex. La porosità dei gel a base di destrano è controllata dalla massa molecolare del destrano usato e dall’ introduzione di legami crociati ottenuti con epicloridrina. Limiti di esclusione kD. Il Sephacryl è destrano con legami crociati ottenuti con N,N’-metilene bisacrilamide. Limiti di esclusione kD. Il Superdex e’ un gel composito costituito da destrano covalentemente legato ad agarosio.")
63
Formazione della matrice

65
Tipi di matrici utilizzate
Poliacrilamide: Polimero di acrilamide e N,N’-metilenbisacrilamide (quest’ultimo determina la formazione di legami crociati). E’ disponibile in commercio come Bio-GelP, con limiti di esclusione tra 0.2 e 400 kD. Polistirene Polimero di stirene legato con divinilbenzene. Silice Polimero prodotto a partire dall’ acido ortosilicico. I molti gruppi silanolo (Si-OH) rendono la matrice altamente idrofilica. Il loro eccesso può essere eliminato derivatizzando con triclorometilsilano.
. E’ disponibile in commercio come Bio-GelP, con limiti di esclusione tra 0.2 e 400 kD. Polistirene Polimero di stirene legato con divinilbenzene. Silice Polimero prodotto a partire dall’ acido ortosilicico. I molti gruppi silanolo (Si-OH) rendono la matrice altamente idrofilica. Il loro eccesso può essere eliminato derivatizzando con triclorometilsilano.")
66
Colonne

67
Così come è importante scegliere l’opportuno materiale di riempimento allo stesso tempo risulta importante selezionare il miglior solvente da impiegare come fase mobile. In genere l’adsorbimento sull’adsorbente avviene molto rapidamente da solventi non polari così questi vengono impiegati per impaccare la colonna e dare inizio allo sviluppo del cromatogramma e come precedentemente sottolineare si può procedere aumentando progressivamente la polarità tenendo conto della serie “eluotropa”. E’ pratica comune introdurre nella colonna concentrazioni crescenti del solvente più polare piuttosto che cambiare bruscamente la composizione del solvente. L’ordine con cui i componenti di una miscela vengono eluiti dalla colonna dipende dalla loro polarità relativa così avendo una miscela contenente due componenti uno più polare rispetto all’altro si rileverà come il meno polare verrà fluito prima con un solvente relativamente non polare e solo successivamente all’aumento della polarità si riscontrerà l’eluizione anche del secondo componente.

68
Serie eluotropa in ordine di polarità crescente
1. Esano, eteri di petrolio 9. Acetato di etile 2. eptano 10. Piridina 3. cicloesano 11. Acetone 4. Tetracloruro di carbonio 12. Propanolo 5. Benzene 13. Etanolo 6. Toluene 14. Metanolo 7. Cloroformio 15. Acqua 8. Etere dietilico 16. Miscele di acidi o di basi con acqua, alcool o piridina

69
L'eluizione può essere effettuata in due modi:
isocratica si mantiene costante la composizione della fase mobile (sia singolo solvente oppure miscela di due o più solventi) per tutta la durata della cromatografia; gradiente si aumenta, durante la cromatografia, la polarità dell'eluente, miscelando solventi diversi. La scelta del solvente o della miscela di solventi é fatta in funzione delle caratteristiche chimico-fisiche delle sostanze da separare.
 per tutta la durata della cromatografia; gradiente. si aumenta, durante la cromatografia, la polarità dell eluente, miscelando solventi diversi. La scelta del solvente o della miscela di solventi é fatta in funzione delle caratteristiche chimico-fisiche delle sostanze da separare.")
70
Strumentazione per HPLC
Un cromatografo HPLC è costituto dalle seguenti parti: riserva di solventi: uno o più solventi che possono essere utilizzati singolarmente o in miscela pompa con pressione fino a 400 atm, flusso stabile tra 0.1 e 10 ml/min sistema di iniezione costituito da una valvola a più vie e da un circuito a volume fisso, o loop, nel quale si mette il campione colonna cromatografica ed eventuale precolonna rivelatore per monitorare gli eluati PC per gestire il sistema e i dati

71
L’HPLC o High Performance Liquid Chromatography ed impiega colonne di acciaio inossidabile. L’uniformità delle dimensioni delle finissime particelle del materiale di riempimento costituisce un fattore particolarmente importante da cui dipende il potere di risoluzione del sistema. I tipi di riempimenti impiegati nell’HPLC possono distinguersi oltre che per le dimensioni oscillanti dai 3 ai 60 m, per la forma regolare, irregolare o sferica e per la tipologia dell’area superficiale:porosa o pellicolare. Così ad esempio riempimenti di diametro 10 m , di forma regolare a corpo poroso presentano come vantaggi l’alta efficienza, una caduta di pressione media ed una capacità da modesta al alta mentre svantaggi sono sicuramente la difficoltà nell’impaccare la colonna. La scelta dei diametri delle colonne dipende da molti fattori. Quanto più sono piccoli , tanto minore sarà la quantità di fase mobile utilizzata, il che permette la rivelazione anche di concentrazioni molto basse di analita.

72
A causa delle pressioni relativamente elevate (tra 1000 e 600 p. s. i
A causa delle pressioni relativamente elevate (tra 1000 e 600 p.s.i.) necessarie per l’esecuzione di separazioni tramite HPLC, in conseguenza delle dimensioni della colonna e della natura fisica del materiale di riempimento l’apparato sperimentale risulta essere piuttosto complesso. Ipotizzando la conduzione di un analisi in gradiente di solvente potremmo così schematizzarlo. Dai due contenitori vengono pompati con velocità di aspirazione programmabile, i solventi da miscelare, questi dopo il mescolamento vengono inviati in una colonna di condizionamento dove incontrano il campione, iniettato attraverso l’iniettore e passano in una pre-colonna che ha funzione di saturare la miscela solvente del liquido che costituisce la fase stazionaria, quindi si ha l’ingresso nella colonna vera e propria a cui è collegato un collettore per la raccolta delle frazioni, un rivelatore ed un registratore.
 necessarie per l’esecuzione di separazioni tramite HPLC, in conseguenza delle dimensioni della colonna e della natura fisica del materiale di riempimento l’apparato sperimentale risulta essere piuttosto complesso. Ipotizzando la conduzione di un analisi in gradiente di solvente potremmo così schematizzarlo. Dai due contenitori vengono pompati con velocità di aspirazione programmabile, i solventi da miscelare, questi dopo il mescolamento vengono inviati in una colonna di condizionamento dove incontrano il campione, iniettato attraverso l’iniettore e passano in una pre-colonna che ha funzione di saturare la miscela solvente del liquido che costituisce la fase stazionaria, quindi si ha l’ingresso nella colonna vera e propria a cui è collegato un collettore per la raccolta delle frazioni, un rivelatore ed un registratore.")
73
Così come nella GC anche l’HPLC necessita di opportuni rivelatori i più utilizzati sono il RID o rivelatore ad indice di rifrazione ed i rivelatori spettroscopici UV. Più ampiamente utilizzati sono gli spettrofotometri che lavorano nel campo dell’ultravioletto e del visibile. Ciascuna sostanza ha un assorbimento caratteristico, cioè ogni composto chimico ha un suo spettro di assorbimento ben definito con bande più o meno intense alle diverse lunghezze d’onda. In cromatografia l’ordinata del cromatogramma rappresenta il segnale quantitativo, poiché più il campione è presente tanto maggiore è l’altezza del picco a parità di condizioni. Poiché i rivelatori UV forniscono un segnale esclusivamente quando si verifica un assorbimento della radiazione che attraversa la cella non tutte le sostanze possono essere rivelate. Le sostanze capaci di assorbire UV sono caratterizzate da uno o più doppi legami, o dalla presenza di doppietti elettronici liberi o composti contenenti gruppi funzionali del tipo: >C=O, >C=S,-N=O,-N=N-

74
Rivelatori per HPLC Bulk properties: si misura una caratteristica della fase mobile che indirettamente rivela gli analiti Solute properties: si misura una caratteristica del soluto QUINDI: Spettrofotometrico UV-visibile: il più comune (quasi tutte le sostanze assorbono nell’UV-vis), misura l’assorbanza dell’eluito a l fissa Spettrofotometrico UV-visibile con Diode-array: misura l’assorbanza dell’eluito in un range di l, restituendo in ogni istante lo spettro UV-vis Spettrofotometrico IR: poco diffuso Spettrofluorimetrico: solo per sostanze che fluorescono (anche con derivatizzazione), molto più sensibile dell’UV-vis a Indice di rifrazione: utilizzato per zuccheri o sostanze non attive nell’UV-vis Elettrochimico: misura la corrente generata ad un elettrodo sul quale avviene una reazione redox che coinvolge l’analita: adatto per sostanze elettroattive, sensibilità eccellente Conducimetrico: misura la corrente trasportata da ioni presenti nell’eluito, utile per sostanze ioniche o ionizzabili Spettrometria di massa: la nuova frontiera, fornisce in ogni istante lo spettro di massa dell’eluito
, misura l’assorbanza dell’eluito a l fissa. Spettrofotometrico UV-visibile con Diode-array: misura l’assorbanza dell’eluito in un range di l, restituendo in ogni istante lo spettro UV-vis. Spettrofotometrico IR: poco diffuso. Spettrofluorimetrico: solo per sostanze che fluorescono (anche con derivatizzazione), molto più sensibile dell’UV-vis. a Indice di rifrazione: utilizzato per zuccheri o sostanze non attive nell’UV-vis. Elettrochimico: misura la corrente generata ad un elettrodo sul quale avviene una reazione redox che coinvolge l’analita: adatto per sostanze elettroattive, sensibilità eccellente. Conducimetrico: misura la corrente trasportata da ioni presenti nell’eluito, utile per sostanze ioniche o ionizzabili. Spettrometria di massa: la nuova frontiera, fornisce in ogni istante lo spettro di massa dell’eluito.")
75
Schema a Blocchi di un Sistema Cromatografico
RIVELATORE

76
Pompa alternativa a pistone
Vantaggi: Robustezza Pressioni in uscita molto elevate (fino a 700 bar) Volume interno ridotto (50 µl) -> adatte all’utilizzo in gradiente Flussi costanti Valvola di ritegno a sfera
 Volume interno ridotto (50 µl) -> adatte all’utilizzo in gradiente. Flussi costanti. Valvola di ritegno a sfera.")
77
Valvole di iniezione L’interfaccia universalmente utilizzata per introdurre il campione è la valvola a 6 (o 7) vie. A seconda del loop montato varia il volume del campione iniettato in colonna

79
High Performance Liquid Chromatography (HPLC)
Elevato numero di piatti teorici Ridotta dimensione delle particelle di matrice Elevata retropressione della colonna

80
Colonne per HPLC Pressione fino a 55 MPa (550 Bar)
")
81
Utilizzabile in gradiente?
Rivelatori Sensibilità adeguata al problema Buona stabilità e riproducibilità Risposta lineare al soluto, possibilmente per parecchi ordini di grandezza Tempi di risposta rapidi Risposta verso tutti i soluti, oppure risposta selettiva verso una o più classi di soluti Rivelatore LOD (ng) Selettività Utilizzabile in gradiente? Assorbimento UV 0.1-1 selettivo SI Indice di rifrazione generale NO Fluorescenza Elettrochimico 0.01-1 Conduttimetrico 0.5-1 Assorbimento IR 1000 Spettrometro di massa
 Selettività. Utilizzabile in gradiente Assorbimento UV selettivo. SI. Indice di rifrazione generale. NO. Fluorescenza Elettrochimico Conduttimetrico Assorbimento IR Spettrometro di massa.")
82
Rivelatore a indice di rifrazione
Il rivelatore a indice di rifrazione misura la differenza nell’indice di rifrazione tra la cella del campione e una cella di riferimento che generalmente contiene soltanto l’eluente. Si utilizza un fascio di luce collimato e filtrato per rimuovere la luce IR che riscalderebbe il campione. Quando l’eluente contenente l’analita entra nella cella del campione, il raggio viene deflesso e inviato al fotodiodo producendo un segnale in uscita differente rispetto a quello prodotto dal solo eluente. Vantaggi e svantaggi È un rivelatore universale, cioè risponde a tutti i composti. Si utilizza per analiti che non assorbono in UV (es. idrocarburi saturi, alcool, eteri) Svantaggi: è poco sensibile, non è compatibile con gradiente di eluizione ed è sensibile a variazioni di p e T.
 Svantaggi: è poco sensibile, non è compatibile con gradiente di eluizione ed è sensibile a variazioni di p e T.")
83
Rivelatore UV a l fissa La radiazione proveniente da una lampada a vapori di Hg passa attraverso la cella del campione e arriva al fotodiodo. L’intensità della luce assorbita è proporzionale alla concentrazione dell’analita. Vantaggi e svantaggi Il principale vantaggio è il basso costo. Inoltre l’elevata intensità della radiazione della lampada a Hg permette di ottenere elevata sensibilità per composti che assorbono a 254 nm. Il principale svantaggio è determinato dalla scarsa selettività dovuta alla necessità di lavorare a l fissa.

84
Il rivelatore UV a l variabile è sicuramente quello maggiormente utilizzato in HPLC.
La luce UV proveniente dalla lampada a D2 e scissa nelle sue componenti attraverso un monocromatore a gradini. L’intensità della luce trasmessa è misurata attraverso un fotodiodo ed è proporzionale alla concentrazione dell’analita Vantaggi Versatilità: possibilità di selezionare l da 190 a 800 nm. Elevata sensibilità: potendo scegliere la l ottimale (max assorbanza) per un analita. Selettività: quando si hanno sovrapposizioni di picchi si può variare la l in modo tale da minimizzare l’assorbimento degli interferenti. Possibilità di utilizzare gradiente di eluizione, scegliendo una l alla quale la miscela solvente non assorbe.
 per un analita. Selettività: quando si hanno sovrapposizioni di picchi si può variare la l in modo tale da minimizzare l’assorbimento degli interferenti. Possibilità di utilizzare gradiente di eluizione, scegliendo una l alla quale la miscela solvente non assorbe.")
85
Rivelatore UV a diode array
Il rivelatore UV a l diode array è quello che attualmente viene sempre più utilizzato in HPLC. La luce UV proveniente dalla lampada a D2 passa attraverso una cella a flusso prima che venga scissa nelle sue componenti attraverso un monocromatore a gradini. L’intensità della luce trasmessa ad ogni l è misurata simultaneamente attraverso un array di alcune centinaia di fotodiodi. Un pc può processare, registrare e mostrare gli spettri in continuo durante l’analisi. Inoltre si possono registrare i cromatogrammi a ciascuna l. Vantaggi e svantaggi Presenta gli stessi vantaggi in termini di versatilità, sensibilità e selettività del rivelatore a l variabile. Fornendo anche gli spettri degli analiti, permette di effettuare anche il riconoscimento dei composti analizzati. Svantaggio: è più costoso rispetto al rivelatore a l variabile.

86
Rivelatore a Fluorescenza
La luce UV proveniente da una lampada (filtrata alla opportuna λ) o da un laser, passa attraverso la cella a flusso. Quando un campione fluorescente passa attraverso la cella, assorbe la radiazione, viene eccitato e quindi emetterà la radiazione di fluorescenza ad una maggiore λ. L’intensità della luce emessa viene misurata attraverso un fotomoltiplicatore posto a 90° rispetto al fascio incidente. Vantaggi e svantaggi È un rivelatore molto sensibile, ma risponde soltanto ai pochi analiti fluorescenti. Per aumentarne l’applicabilità si possono legare covalentemente dei marker fluorescenti. Questa derivatizzazione può essere fatta o prima della separazione o post-colonna aggiungendo i reattivi marcanti tra la colonna e il rivelatore.
 o da un laser, passa attraverso la cella a flusso. Quando un campione fluorescente passa attraverso la cella, assorbe la radiazione, viene eccitato e quindi emetterà la radiazione di fluorescenza ad una maggiore λ. L’intensità della luce emessa viene misurata attraverso un fotomoltiplicatore posto a 90° rispetto al fascio incidente. Vantaggi e svantaggi. È un rivelatore molto sensibile, ma risponde soltanto ai pochi analiti fluorescenti. Per aumentarne l’applicabilità si possono legare covalentemente dei marker fluorescenti. Questa derivatizzazione può essere fatta o prima della separazione o post-colonna aggiungendo i reattivi marcanti tra la colonna e il rivelatore.")
87
Cromatografie di ripartizione
Fase diretta (fase staz. polare, eluente apolare) Fase inversa (fase staz. apolare, eluente polare) Si – O – C18H37
 Fase inversa (fase staz. apolare, eluente polare) Si – O – C18H37.")
88
RPC – Eluizione in gradiente acqua – CH3CN

89
Cromatografie di adsorbimento
Interazione idrofobica Scambio ionico Per affinità Affinità per ligandi (anticorpi) Proteine ingegnerizzate (His-tag) Immobilized Metal ion Affinity Chromatography
 Proteine ingegnerizzate (His-tag) Immobilized Metal ion Affinity Chromatography.")
90
Coefficiente di partizione - a
Il coefficiente di partizione tra le due fasi, a, è definito come il rapporto tra il soluto adsorbito sulla fase stazionaria rispetto a quello in soluzione nella fase mobile. a = 0 nessun adsorbimento a = 1 tutto adsorbito

91
Cromatografia di scambio ionico
Scambio anionico A pH maggiore del pI (almeno una unità) la proteina è carica negativamente e lega la colonna a scambio anionico. L’eluizione avviene con [Cl-] crescente o abbassando il pH (più difficile da controllare). Scambio cationico A pH minore del pI (almeno una unità) la proteina è carica positivamente e lega la colonna a scambio cationico. L’eluizione avviene con [Na+] crescente o alzando il pH.
 la proteina è carica negativamente e lega la colonna a scambio anionico. L’eluizione avviene con [Cl-] crescente o abbassando il pH (più difficile da controllare). Scambio cationico. A pH minore del pI (almeno una unità) la proteina è carica positivamente e lega la colonna a scambio cationico. L’eluizione avviene con [Na+] crescente o alzando il pH.")
92
Scambiatori ionici

93
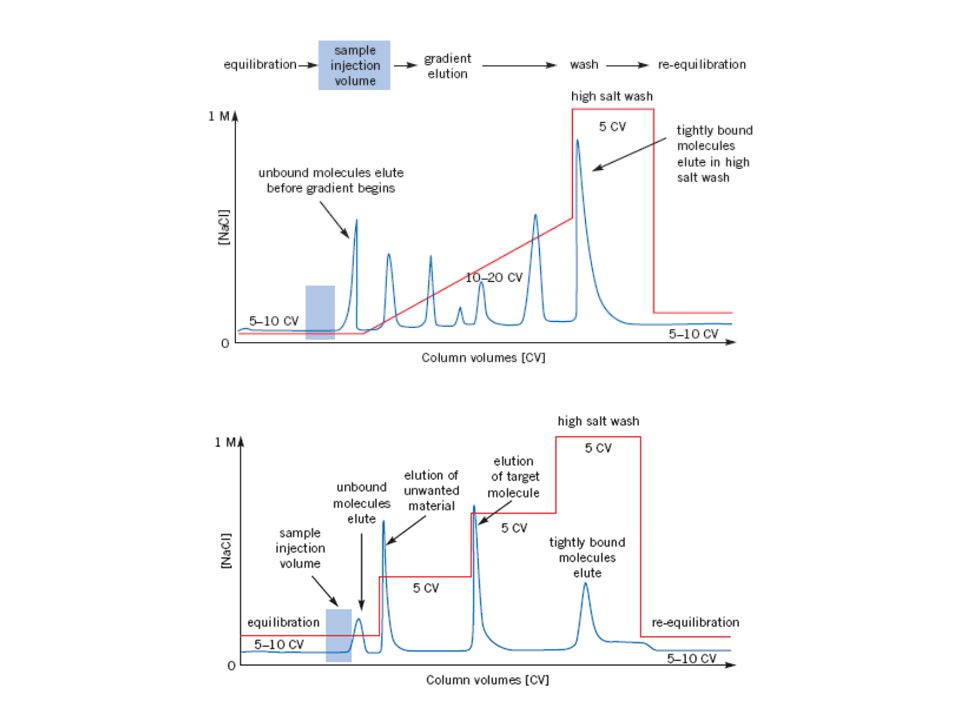
Cromatografia di scambio ionico

96
Cromatografia di scambio ionico
VANTAGGI Per grandi volumi, Grandi quantità di proteina (1-5 g proteina per 100 ml), Matrice molto robusta, Condizioni molto flessibili, possibili molte variazioni. SVANTAGGI Proteine allo stesso pH e concentrazioni di sali della colonna. Ciò può essere scomodo poiché poi occorre passare in dialisi per togliere i sali.
, Matrice molto robusta, Condizioni molto flessibili, possibili molte variazioni. SVANTAGGI. Proteine allo stesso pH e concentrazioni di sali della colonna. Ciò può essere scomodo poiché poi occorre passare in dialisi per togliere i sali.")
97
Gas cromatografia La gascromatografia è impiegata per la separazione di sostanze volatili. Si presta meno facilmente a misure quantitative rispetto alla LC, in compenso ha maggiori potenzialità dal punto di vista diagnostico. Si possono separare sostanze appartenenti a varie classi tra cui, di interesse archeometrico: aromi (terpeni, esteri) idrocarburi a catena corta acidi carbossilici composti di interesse biochimico
 idrocarburi a catena corta. acidi carbossilici. composti di interesse biochimico.")
98
Nella gascromatografia il campione è vaporizzato e poi iniettato in colonna; un gas costituisce la fase mobile ma in questo caso non ha alcuna interazione con i soluti in quanto agisce soltanto da carrier, cioè trasporta i soluti lungo la colonna I composti iniettabili in un sistema GC devono avere Teb < 300°C e non devono essere termolabili, ovvero non devono degradarsi per effetto della temperatura, pena l’impossibilità di riconoscerli nel campione

99
Gascromatografia Gas-liquido Gas-solido supporto inerte solido
liquido non volatile, legato covalentemente meccanismo di ripartizione moltissime applicazioni Gas-solido fasi stazionarie di silice, allumina o carbone meccanismo di adsorbimento adatta per la separazione di gas permanenti (H2, He, Ar, O2, N2, CO) o idrocarburi a basso punto di ebollizione
 o idrocarburi a basso punto di ebollizione.")
100
Cromatografia liquida
Riassumendo analiti volatili o volatilizzabili, termicamente stabili, non ionici Gascromatografia analiti non volatili o poco volatili, ionici, ionizzabili o non ionici, termicamente instabili Cromatografia liquida

101
Schema a blocchi di un gascromatografo

102
Schema di iniettore L’apparato di iniezione è costituito da una camera metallica che viene portata ad una temperatura di circa 50°C più elevata rispetto alla temperatura del composto meno volatile presente nel campione. A questa temperatura il campione viene istantaneamente vaporizzato e viene portato dal carrier gas in cima alla colonna.

103
Colonna Le colonne capillari hanno diametri piuttosto piccoli e sono piuttosto lunghe e la loro forma può essere ad U oppure a spirale. I vantaggi di impiego delle colonne capillari dipendono dal fatto che sono estremamente ridotti fenomeni di diffusione, a tutto vantaggio della efficienza separativa: queste presentano la fase stazionaria come uno strato sottile aderente alla parete del tubo, oppure con la fase liquida depositata su uno strato sottile di supporto solido.

104
Rivelatore a ionizzazione di fiamma

105
Accoppiamento di gascromatografo con spettrometro di massa (sorgente a ionizzazione diretta)
")
106
La colonna di un GC può essere mantenuta sia in condizioni isoterme che di gradiente di temperatura o alternare condizioni di isoterme a gradienti di temperatura. Le migliori condizioni di analisi in termini di temperatura corrispondono a temperature uguali o di poco superiori al punto di ebollizione medio del campione consentendo l’ottenimento di buoni tempi di ritenzione. GC/MS

107
Spettro di massa dell’alcaloide senecifillina

108
Il grosso limite della GC risiede nel fatto che composti ad elevato peso molecolare, difficilmente vaporizzabili, oppure esprimenti gruppi funzionali (carbossilici, amminici primari, ecc.) che possono interagire in maniera non-ideale con la fase stazionaria devono essere precedentemente sottoposti a pirolisi o a derivatizzazione tipo esterificazione o trattamento con derivati del silicio. I limiti della derivatizzazione sono di due ordini: a seguito di tale procedura non è più possibile operare analisi di tipo quantitativo né si può essere certi di non aver alterato le strutture dell’analita in esame. Recentemente le convenzionali tecniche cromatografiche sono state modificate in modo tale che possano eseguirsi analisi di miscele di composti solidi con una risoluzione paragonabili alla GLC. Questa nuova tecnica è particolarmente idonea all’analisi di composti non volatili o termicamente instabili al di fuori della portata della GLC.

Presentazioni simili












>")




>")





 Isolamento e Purificazione dei Composti Organici>")