Scaricare la presentazione
1
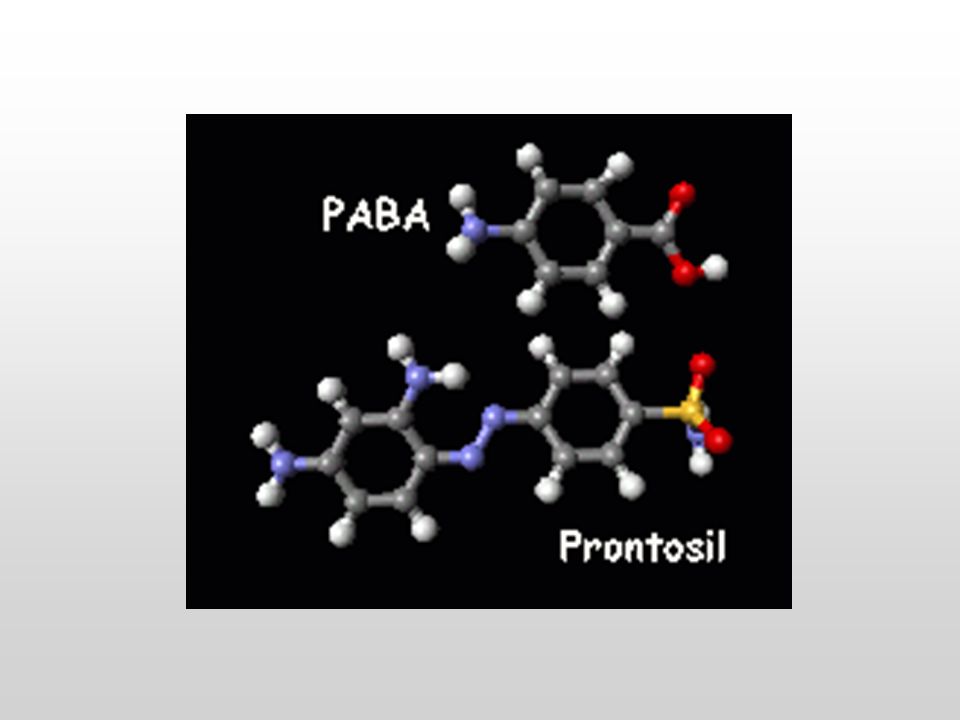
Gerhard Johannes Paul Domagk (1895-1964)
")
5
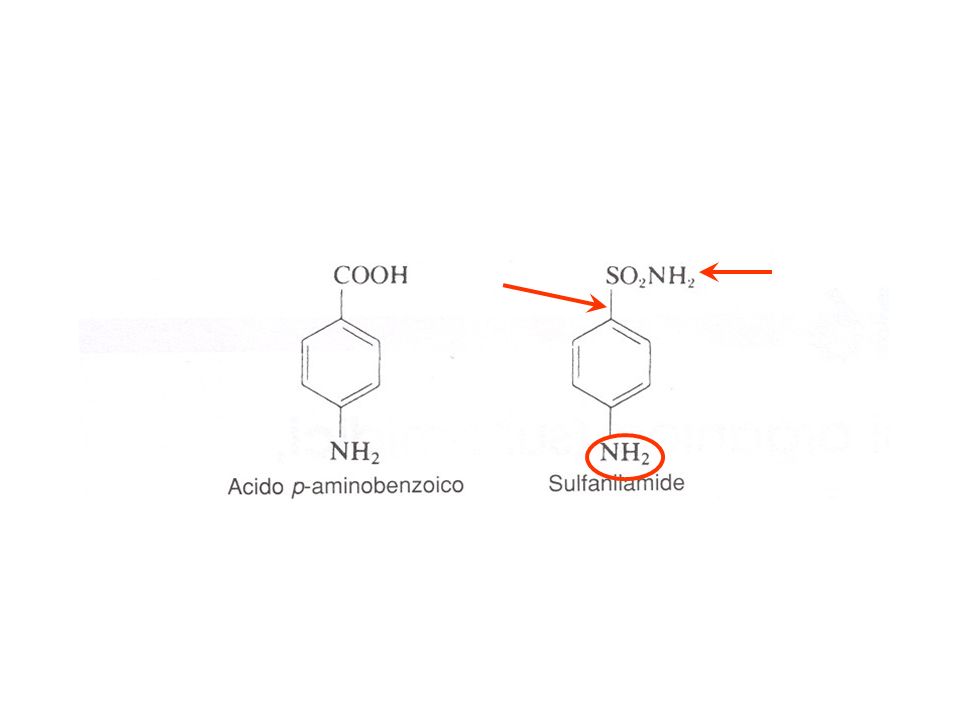
BIOSINTESI DELL’ACIDO FOLICO

6
BIOSINTESI DELLE BASI PURINICHE
METENILTETRAIDROFOLATO N10-FORMILTETRAIDROFOLATO

7
BIOSINTESI DEL TIMIDILATO

8
SULFAMIDICI SISTEMICI AD AZIONE RAPIDA

9
SULFAMIDICI SISTEMICI SEMI-RETARD

10
SULFAMIDICI ATTIVI NEL LUME INTESTINALE
La sulfasalazina è poco assorbita dall’intestino. Utilizzata nella colite ulcerosa Sulfasalazina Sulfapiridina (responsabile di buona parte degli effetti collaterali) 5-ASA (acido 5-aminosalicilico o mesalamina; azione anti-infiammatoria
 5-ASA. (acido 5-aminosalicilico o mesalamina; azione anti-infiammatoria.")
11
SULFAMIDICI PER USO TOPICO

12
TRIMETOPRIM

13
PIRIMETAMINA

14
SELETTIVITA’ DEGLI INIBITORI DELLA DIIDROFOLATO REDUTTASI (FH2)
INIBITORE IC50 (mol/l) per FH2 reduttasi UOMO PROTOZOI BATTERI TRIMETOPRIM 260 0.07 0.005 PIRIMETAMINA 0.0005 2.5 METOTREXATO 0.001 0.1 inattivo
 per FH2 reduttasi. UOMO. PROTOZOI. BATTERI. TRIMETOPRIM PIRIMETAMINA METOTREXATO 0.1. inattivo.")
15
COMBINAZIONI SINERGICHE DEI SULFAMIDICI
+ = COTRIMOSSAZOLO (BACTRIM®) H. influenzae, P. carinii, Enterobacteriaceae
 H. influenzae, P. carinii, Enterobacteriaceae.")
16
COMBINAZIONI SINERGICHE DEI SULFAMIDICI
Sulfadoxina + = FANSIDAR P. falciparum

17
COMBINAZIONI SINERGICHE DEI SULFAMIDICI
+ Toxoplasma gondii

18
MECCANISMI DI RESISTENZA AI SULFAMIDICI
1) permeabilità batterica o estrusione attiva del farmaco 2) produzione di un metabolita essenziale o di un antagonista del farmaco 3) affinità della diidropteroato sintetasi nei confronti dei SA 4) attivazione di una via metabolica alternativa per la sintesi di un metabolita essenziale
 permeabilità batterica o estrusione attiva del. farmaco. 2) produzione di un metabolita essenziale o di un. antagonista del farmaco. 3) affinità della diidropteroato sintetasi nei. confronti dei SA. 4) attivazione di una via metabolica alternativa per. la sintesi di un metabolita essenziale.")
19
EFFETTI COLLATERALI DEI SULFAMIDICI
Disturbi del tratto urinario; cristalluria Disordini del sistema emopoietico: agranulocitosi anemia aplastica anemia emolitica acuta

21
EFFETTI COLLATERALI DEI SULFAMIDICI
Disturbi del tratto urinario; cristalluria Disordini del sistema emopoietico: agranulocitosi anemia aplastica anemia emolitica acuta kernicterus nel neonato Reazioni di ipersensibilità Il kernicterus nel neonato è dovuto a un accumulo di bilirubina nel sistema nervoso centrale. La bilirubina è prodotta durante il catabolismo dell’eme a partire dalla biliverdina ad opera della eme-ossigenasi. La bilirubina ha natura lipofila ed è trasportata nel plasma legata all’albumina. In questa forma non è in grado di attraversare la BBB, e viene trasportata al fegato dove viene coniugata all’ac. Glicuronico da p. della UDPGT.. La bilirubina coniugata viene escreta nell’intestino per via biliare. Qui la flora intestinale la trasforma in urobilinogeno che viene eliminato. Nel neonato diversi fattori contribuiscono a una condizione transitoria di iperbilirubinemia, alcuni dei quali legati ad eventi fisiologici avversi. 1) aumento della produzione (i neonati hanno di per sé una produzione doppia rispetto all’adulto, principalmente per un maggiore volume circolare di RBC che hanno una durata di vita breve.(a) Fattori prenatali, come l’abitudine al fumo o malattie materne, insufficienza placentare o gravidanza condotta ad altitudine elevata, o accidenti legati al parto possono portare a policitemia (aumento della massa di RBC). (b) L’emolisi su base immunitaria, specialmente legata a incompatibilità Rh, o dovuta ad anomalie dei RBC è spesso responsabile di iperbilirubinemia nel neonato. I difetti dei RBC si classificano in difetti di membrana (sferocitosi o elliptocitosi erediarie), difetti enzimatici (e.g. deficit di G6PDH) e emoglobinopatie (alfa e beta talassemia). (c) stravaso di sangue (ecchimosi prodotte da traumi durante il parto o emorragie interne). (d) induzione della eme ossigenasi-1 a causa di stress quali acidosi, ipossia, ipotermia o infezioni. (e) fattori epidemiologici (maschi > femmine etc.) 2) ridotta eliminazione. (a) difetti nel trasporto. L’abumina ha un sito ad alta affinità e due siti a bassa affinità per la bilirubina. Nel neonato l’affinità di legame dell’albumina per la bilirubina è bassa e questo ne compromette il trasporto al fegato e l’eliminazione, oltre a indurre un aumento dei livelli di bilirubina libera in grado di attraversare la BBB. La capacità di legame dell’albumina per la bilirubina può essere ridotta da diversi farmaci, quali p.e. sulfamidici e alcune cefalosporine. (b) uptake e coniugazione a livello epatico. Nel neonato fino a 5 gg i livelli di ligandina (proteina accettrice della bilirubina) sono molto bassi. Inoltre, nel neonato e nei primi 3-4 mesi di vita la UDPGT non è pienamente funzionale; durante i primi dieci gg i livelli di attività sono circa lo 0.1% rispetto a quelli dell’adulto, il che rappresenta la causa principale dell’ittero fisiologico del neonato. Difetti congeniti ereditari del gene per la UDPGT possono dare luogo a sindromi di gravità variabile, come la sindrome di Crigler-Najjar (che comporta il rischio di kernicterus per tutta la vita) e la malattia di Gilbert (legata alla presenza di una tripletta ripetuta).(c) Il neonato è inizialmente sprovvisto di flora batterica intestinale in grado di trasformare la bilirubina coniugata in urobilinogeno. In compenso nell’intestino del neonato, ma non nell’adulto, sono presenti beta-glicuronidasi in grado di deconiugare la bilirubina e di dare luogo a un circolo enteroepatico, favorito anche dall’iniziale lentezza del transito intestinale nel neonato. 3) esistono fattori sistemici che aumentano il rischio di kernicterus in assenza di iperbilirubinemia grave. (a) galattosemia. (b) ipotiroidismo. (c) farmaci (vedere sopra). (d) acidosi. L’acidosi sistemica riduce l’affinità dell’albumina per la bilirubina; inoltre favorisce la formazione di bilirubina acida il cui uptake a livello del sistema nervoso è facilitato. (d) alterazioni della BBB, già di per sé meno efficace nel neonato, a causa di infezioni o altre condizioni Nel neonato e nei primi 3-4 mesi di vita questo enzima non è pienamente funzionale Nel neonato la bilirubina viene deconiugata da una bete-glicuronidasi intestinale, il che dà luogo a un ricircolo entero-epatico, tipico del neonato
 aumento della produzione (i neonati hanno di per sé una produzione doppia rispetto all’adulto, principalmente per un maggiore volume circolare di RBC che hanno una durata di vita breve.(a) Fattori prenatali, come l’abitudine al fumo o malattie materne, insufficienza placentare o gravidanza condotta ad altitudine elevata, o accidenti legati al parto possono portare a policitemia (aumento della massa di RBC). (b) L’emolisi su base immunitaria, specialmente legata a incompatibilità Rh, o dovuta ad anomalie dei RBC è spesso responsabile di iperbilirubinemia nel neonato. I difetti dei RBC si classificano in difetti di membrana (sferocitosi o elliptocitosi erediarie), difetti enzimatici (e.g. deficit di G6PDH) e emoglobinopatie (alfa e beta talassemia). (c) stravaso di sangue (ecchimosi prodotte da traumi durante il parto o emorragie interne). (d) induzione della eme ossigenasi-1 a causa di stress quali acidosi, ipossia, ipotermia o infezioni. (e) fattori epidemiologici (maschi > femmine etc.) 2) ridotta eliminazione. (a) difetti nel trasporto. L’abumina ha un sito ad alta affinità e due siti a bassa affinità per la bilirubina. Nel neonato l’affinità di legame dell’albumina per la bilirubina è bassa e questo ne compromette il trasporto al fegato e l’eliminazione, oltre a indurre un aumento dei livelli di bilirubina libera in grado di attraversare la BBB. La capacità di legame dell’albumina per la bilirubina può essere ridotta da diversi farmaci, quali p.e. sulfamidici e alcune cefalosporine. (b) uptake e coniugazione a livello epatico. Nel neonato fino a 5 gg i livelli di ligandina (proteina accettrice della bilirubina) sono molto bassi. Inoltre, nel neonato e nei primi 3-4 mesi di vita la UDPGT non è pienamente funzionale; durante i primi dieci gg i livelli di attività sono circa lo 0.1% rispetto a quelli dell’adulto, il che rappresenta la causa principale dell’ittero fisiologico del neonato. Difetti congeniti ereditari del gene per la UDPGT possono dare luogo a sindromi di gravità variabile, come la sindrome di Crigler-Najjar (che comporta il rischio di kernicterus per tutta la vita) e la malattia di Gilbert (legata alla presenza di una tripletta ripetuta).(c) Il neonato è inizialmente sprovvisto di flora batterica intestinale in grado di trasformare la bilirubina coniugata in urobilinogeno. In compenso nell’intestino del neonato, ma non nell’adulto, sono presenti beta-glicuronidasi in grado di deconiugare la bilirubina e di dare luogo a un circolo enteroepatico, favorito anche dall’iniziale lentezza del transito intestinale nel neonato. 3) esistono fattori sistemici che aumentano il rischio di kernicterus in assenza di iperbilirubinemia grave. (a) galattosemia. (b) ipotiroidismo. (c) farmaci (vedere sopra). (d) acidosi. L’acidosi sistemica riduce l’affinità dell’albumina per la bilirubina; inoltre favorisce la formazione di bilirubina acida il cui uptake a livello del sistema nervoso è facilitato. (d) alterazioni della BBB, già di per sé meno efficace nel neonato, a causa di infezioni o altre condizioni. Nel neonato e nei primi 3-4 mesi di vita questo enzima non è pienamente funzionale. Nel neonato la bilirubina viene deconiugata da una bete-glicuronidasi intestinale, il che dà luogo a un ricircolo entero-epatico, tipico del neonato.")
22
SINDROME DI STEVENS-JOHNSON/SINDROME DI LYELL

23
POSSIBILE IPERSENSIBILITA’ CROCIATA
NEI CONFRONTI DI SULFONILUREE DIURETICI acetazolamide diuretici tiazidici diuretici dell’ansa

24
STRUTTURA DEI SULFONI

25
COMBINAZIONI SINERGICHE DEI SULFONI
Cl Sulfadoxina + Chlorproguanil = LAPDAP P. falciparum

26
CHINOLONI E FLUOROCHINOLONI

27
BERSAGLI MOLECOLARI DEI CHINOLONI
E FLUOROCHINOLONI DNA Girasi Topoisomerasi IV Scoperta nel 1976 Codificata da gyrA e gyrB Superavvolgimento di DNA rilassato Bersaglio principale nei batteri Gram - Scoperta nel 1990 Codificata da parC e parE Decatenazione delle molecole figlie di DNA Bersaglio principale nei batteri Gram +

28
Interruption of gyrase action by quinolones
Interruption of gyrase action by quinolones. (a) DNA gyrase and DNA before strand passage. Gyrase, represented schematically by thin crossed lines, binds DNA. (b) Opening of the DNA gate. Gyrase undergoes a conformational change, DNA is broken as a pair of staggered single-strand breaks, and another region of the same DNA molecule, shown in cross section, is brought close to the DNA gate. Quinolones trap gyrase at this stage. (c) Strand passage. The region of DNA shown in cross section passes through the DNA gate. (d) DNA gyrase and DNA after strand passage. The reactions are shown as reversible because gyrase can introduce and remove negative supercoils from DNA. The quinolones block both reactions. High [ATP]/[ADP] ratios drive the reaction to the right in the supercoiling direction, and coumarin antibiotics or low [ATP]/[ADP] ratios cause relaxation. Adapted from reference 23 with permission.
 DNA gyrase and DNA before strand passage. Gyrase, represented schematically by thin crossed lines, binds DNA. (b) Opening of the DNA gate. Gyrase undergoes a conformational change, DNA is broken as a pair of staggered single-strand breaks, and another region of the same DNA molecule, shown in cross section, is brought close to the DNA gate. Quinolones trap gyrase at this stage. (c) Strand passage. The region of DNA shown in cross section passes through the DNA gate. (d) DNA gyrase and DNA after strand passage. The reactions are shown as reversible because gyrase can introduce and remove negative supercoils from DNA. The quinolones block both reactions. High [ATP]/[ADP] ratios drive the reaction to the right in the supercoiling direction, and. coumarin antibiotics or low [ATP]/[ADP] ratios cause relaxation. Adapted from reference 23 with permission.")
29
K. Drlica et al., Antimicrob. Agents Chemother. 52: 385-392, 2008

31
Intracellular action of quinolones
Intracellular action of quinolones. (a) DNA gyrase and DNA interact to form a cleaved complex. (b) Quinolones trap the cleaved complex. Gyrase mutations prevent trapping by the quinolones. The trapped complexes block DNA synthesis and cell growth. (c) A putative trapped complex removal system releases lethal double-strand DNA breaks from the complexes. Chloramphenicol (CAM) or rifampin treatment of cells blocks this reaction. (d) Fluoroquinolones at high concentration stimulate gyrase subunit dissociation, which releases lethal double-strand breaks. The dotted line indicates release of staggered doublestrand DNA breaks when cell lysates are treated with ionic detergents such as sodium dodecyl sulfate. KARL DRLICA* AND XILIN ZHAO: MICROBIOLOGY AND MOLECULAR BIOLOGY REVIEWS, Sept. 1997, p. 377–392
 DNA gyrase and DNA interact to form a cleaved complex. (b) Quinolones trap the cleaved complex. Gyrase mutations prevent trapping by the quinolones. The trapped complexes block DNA synthesis and cell growth. (c) A putative trapped complex removal system releases lethal double-strand DNA breaks from the complexes. Chloramphenicol (CAM) or rifampin treatment of cells blocks this reaction. (d) Fluoroquinolones at high concentration stimulate gyrase subunit dissociation, which releases lethal. double-strand breaks. The dotted line indicates release of staggered doublestrand DNA breaks when cell lysates are treated with ionic detergents such as sodium dodecyl sulfate. KARL DRLICA* AND XILIN ZHAO: MICROBIOLOGY AND MOLECULAR BIOLOGY REVIEWS, Sept. 1997, p. 377–392.")
32
EFFETTI COLLATERALI DEI FLUOROCHINOLONI
Effetti gastrointestinali Effetti sul SNC Reazioni di ipersensibilità e fotosensibilizzazione Disordini ematologici Epatotossicità (trovafloxacina)
")
33
MECCANISMI MOLECOLARI DI RESISTENZA AI CHINOLONI E FLUOROCHINOLONI
CROMOSOMICI Mutazioni in geni che codificano per le subunità della DNA girasi (o della topoisomerasi IV) Diminuzione della permeabilità di membrane Aumentato efflusso del farmaco MEDIATI DA PLASMIDI Espressione di proteine Qnr
 Diminuzione della permeabilità di membrane. Aumentato efflusso del farmaco. MEDIATI DA PLASMIDI. Espressione di proteine Qnr.")
34
RESISTENZA AI CHINOLONI MEDIATA
DA PLASMIDI (PMQR) In wild-type strains, resistance was low, but in certain clinical isolates deficient in outer membrane porins the plasmid raised quinolone resistance to more than 256 µg/mL. Plasmid-mediated quinolone resistance also facilitated selection of higher quinolone resistance. The low resistance is thought to allow the bacterial population to reach a concentration at which secondary mutations to higher resistance can occur. Streptomycin and quinolone resistance are carried by plasmid pMG252, and the frequency of mutations to either higher streptomycin or quinolone resistance was increased in E coli J53 with pMG252, compared with the plasmid-free parent. The mutations that increased quinolone resistance occurred on the E coli chromosome and have not yet been mapped; however, because they can be selected by stepwise increases in quinolone concentration and seem to affect susceptibility to cefoxitin, chloramphenicol, and tetracycline, they may arise at the multiple antibiotic resistance (mar) locus. The designation qnr is proposed for the plasmid mediated quinolone-resistance locus. Robicsek et al., Lancet Infect Dis 6, 629–640, 2006
 In wild-type strains, resistance was low, but in certain clinical isolates deficient in outer membrane porins the plasmid raised quinolone resistance to more than 256 µg/mL. Plasmid-mediated quinolone resistance also facilitated selection of higher quinolone resistance. The low resistance is thought to allow the bacterial population to reach a concentration at which secondary mutations to higher resistance can occur. Streptomycin and quinolone resistance are carried by plasmid pMG252, and the frequency of mutations to either higher streptomycin or quinolone resistance was increased in E coli J53 with pMG252, compared with the plasmid-free parent. The mutations that increased quinolone resistance occurred on the E coli chromosome and have not yet been mapped; however, because they can be selected by stepwise increases in quinolone concentration and seem to affect susceptibility to cefoxitin, chloramphenicol, and tetracycline, they may arise at the multiple antibiotic resistance (mar) locus. The designation qnr is proposed for the plasmid mediated quinolone-resistance locus. Robicsek et al., Lancet Infect Dis 6, 629–640,")
35
MECCANISMI MOLECOLARI DI RESISTENZA AI CHINOLONI E FLUOROCHINOLONI
CROMOSOMICI Mutazioni in geni che codificano per le subunità della DNA girasi (o della topoisomerasi IV) Diminuzione della permeabilità di membrane Aumentato efflusso del farmaco MEDIATI DA PLASMIDI Espressione di proteine Qnr Espressione di una aminoglicoside acetiltransferasi fluorochinolone-specifica
 Diminuzione della permeabilità di membrane. Aumentato efflusso del farmaco. MEDIATI DA PLASMIDI. Espressione di proteine Qnr. Espressione di una aminoglicoside acetiltransferasi fluorochinolone-specifica.")
36
RESISTENZA AI CHINOLONI MEDIATA
DA PLASMIDI (PMQR)
")
37
NORFLOXACIN MIC = (0.625) µM CIPROFLOXACIN MIC = 0.02 ( ) µM LEVOFLOXACIN MIC = 0.08 (0.08) µM GEMIFLOXACIN MIC = (0.005) µM
 µM.")
38
MECCANISMI MOLECOLARI DI RESISTENZA AI CHINOLONI E FLUOROCHINOLONI
CROMOSOMICI Mutazioni in geni che codificano per le subunità della DNA girasi (o della topoisomerasi IV) Diminuzione della permeabilità di membrane Aumentato efflusso del farmaco MEDIATI DA PLASMIDI Espressione di proteine Qnr Espressione di una aminoglicoside acetiltransferasi fluorochinolone-specifica Espressione di un trasportatore della famiglia MFS
 Diminuzione della permeabilità di membrane. Aumentato efflusso del farmaco. MEDIATI DA PLASMIDI. Espressione di proteine Qnr. Espressione di una aminoglicoside acetiltransferasi fluorochinolone-specifica. Espressione di un trasportatore della famiglia MFS.")
39
RESISTENZA AI CHINOLONI MEDIATA
DA PLASMIDI (PMQR)
")
40
STRUTTURA DEI NITROIMIDAZOLI
Attivo su organismi anaerobi Meccanismo d’azione mediato dalla riduzione da parte di proteine contenenti clusters Fe-S (ferredossine)
")
41
ATTIVAZIONE DEL METRONIDAZOLO
Metronidazole is a prodrug: it contains a nitro group that must be reduced for the drug to become active. Reduced metronidazole is highly effective against anaerobic organisms, probably because of the formation of cytotoxic intermediates that cause DNA, protein and membrane damage. Two aspects of anaerobic metabolism provide opportunities for selective reduction of the nitro group. First, the reaction catalyzed by PFOR results in the reduction of ferredoxin; reduced ferredoxin can then transfer its electrons to metronidazole, resulting in reduced (active) metronidazole and re-oxidized ferredoxin. Second, many anaerobic organisms express nitroreductase enzymes that selectively reduce metronidazole and, in the process, oxidize NADPH to NADP+
 metronidazole and re-oxidized ferredoxin. Second, many anaerobic organisms express nitroreductase enzymes that selectively reduce metronidazole and, in the process, oxidize NADPH to NADP+")