Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Le malattie ereditarie e i loro meccanismi molecolari

3
Gene Trascrizione Proteine Sintesi proteca “splicing” del mRNA enzimi
recettori trasportatori strutturali trasmettitori passaggio nel citoplasma Sintesi proteca

4
Replicazione Mitosi 2n 2n 4n 4n 2n

5
Continuamente a partire dalla maturità sessuale
Meiosi Alcuni milioni nelle ovaie alla nascita Bloccati all’inizio della I divisione meiotica Solo alcune centinaia matureranno Continuamente a partire dalla maturità sessuale Completamento della II divisione meiotica solo in caso di fertilizzazione da parte di spermatozoo A partire dalla maturità sessuale cicli di ovulazione con completamento della I divisione meiotica e ingresso in II divisione meiotica

6
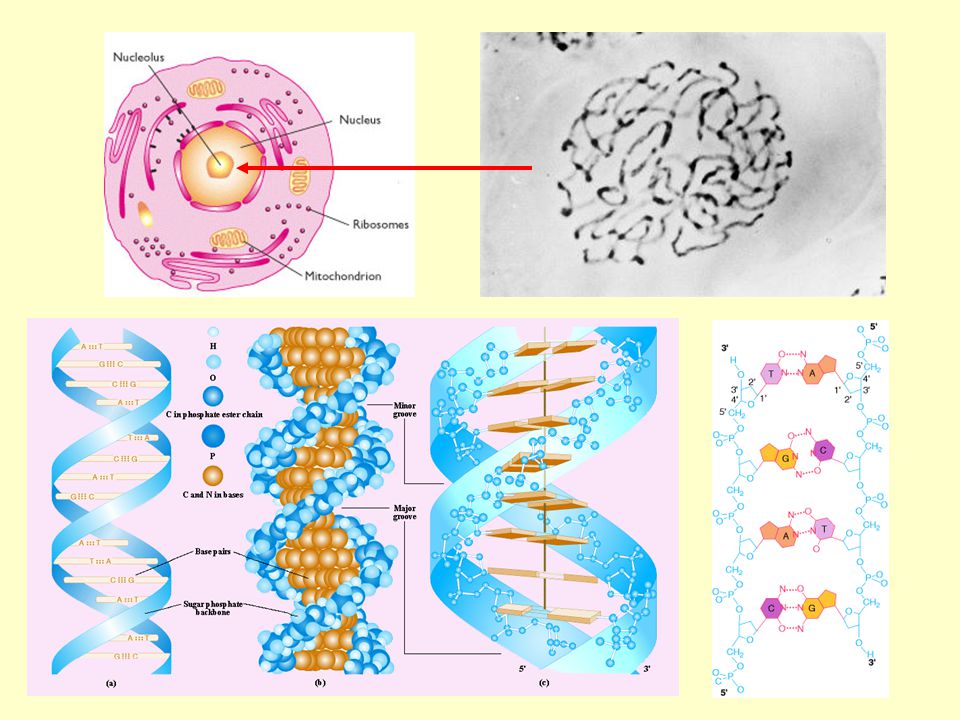
Gene Sequenza di DNA cromosomico contenente le informazioni per la sintesi di un prodotto funzionale Locus Posizione fisica di un determinata sequenza di DNA sul cromosoma Polimorfismo Presenza, in individui diversi, di sequenze di DNA differenti in un determinato locus Allele Una delle possibili varianti alternative con cui un polimorfismo si presenta nella popolazione Genotipo Combinazione di alleli paterni e materni presenti ad un determinato locus in cromosomi omologhi Omozigote / Eterozigote Soggetto con genotipo caratterizzato da alleli paterni e materni identici / diversi Fenotipo Espressione visibile dell’azione di un gene; il quadro clinico di una malattia genetica Mutazione Cambiamento permanente in una sequenza di DNA, indotto da errori nel processo di replicazione del DNA; se interessa le cellule germinali, la mutazione verrà ereditata dalla progenie Test genetico “Analisi di specifici geni, dei loro prodotti o della loro funzione, nonché ogni altro tipo di indagine del DNA o dei cromosomi, finalizzate ad individuare o a escludere modificazioni del DNA verosimilmente associate a patologie genetiche” Comitato nazionale di Bioetica, 1999

7
Il patrimonio genetico della specie umana va incontro a continue modificazioni; questo salvaguarda la capacità di adattamento all’ambiente. Tuttavia, alcune di queste modificazioni, o mutazioni, comportano malattie genetiche.

8
Esistono 4 gruppi principali di patologie che possono interessare i geni:
1. Anomalie cromosomiche 2. Malattie geniche (tra cui rientrano gli errori congeniti del metabolismo e le emoglobinopatie) 3. Malformazioni congenite 4. Infezioni fetali.
 3. Malformazioni congenite. 4. Infezioni fetali.")
9
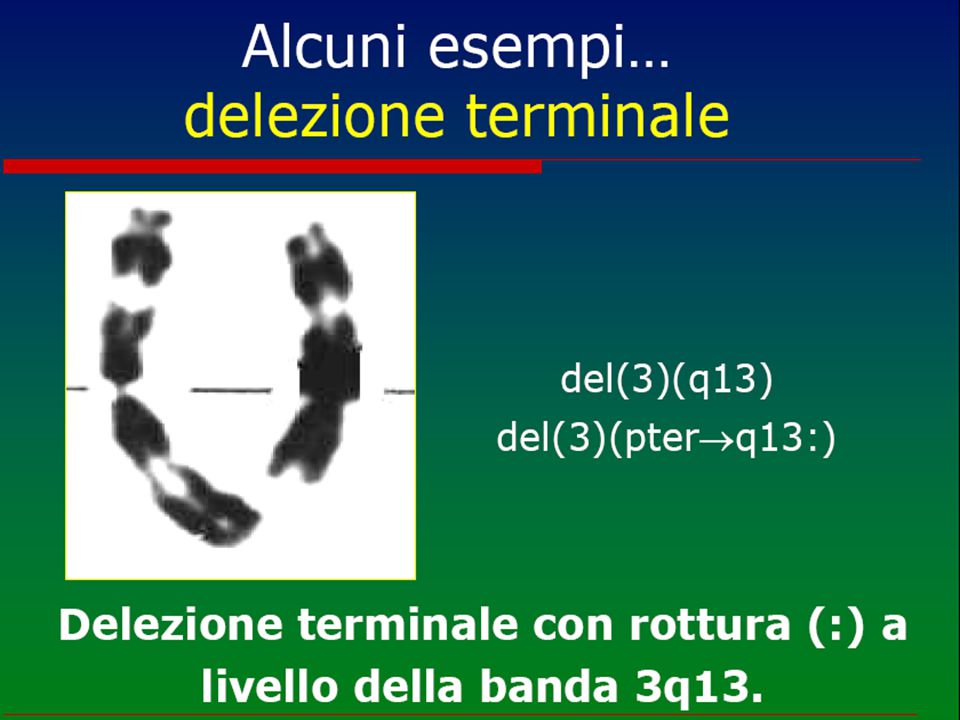
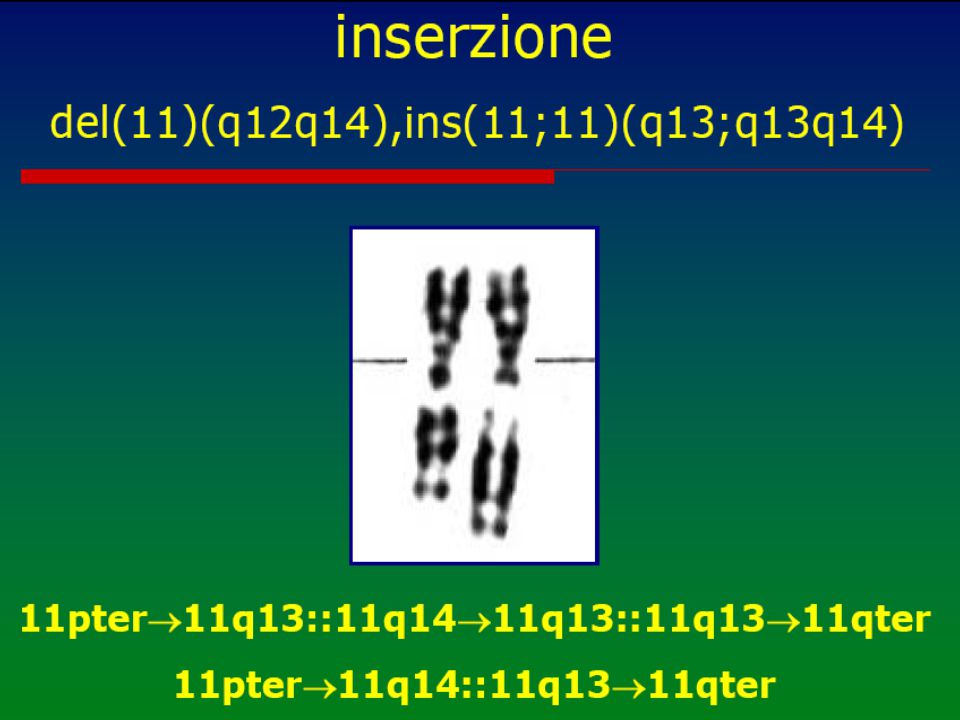
Anomalie cromosomiche
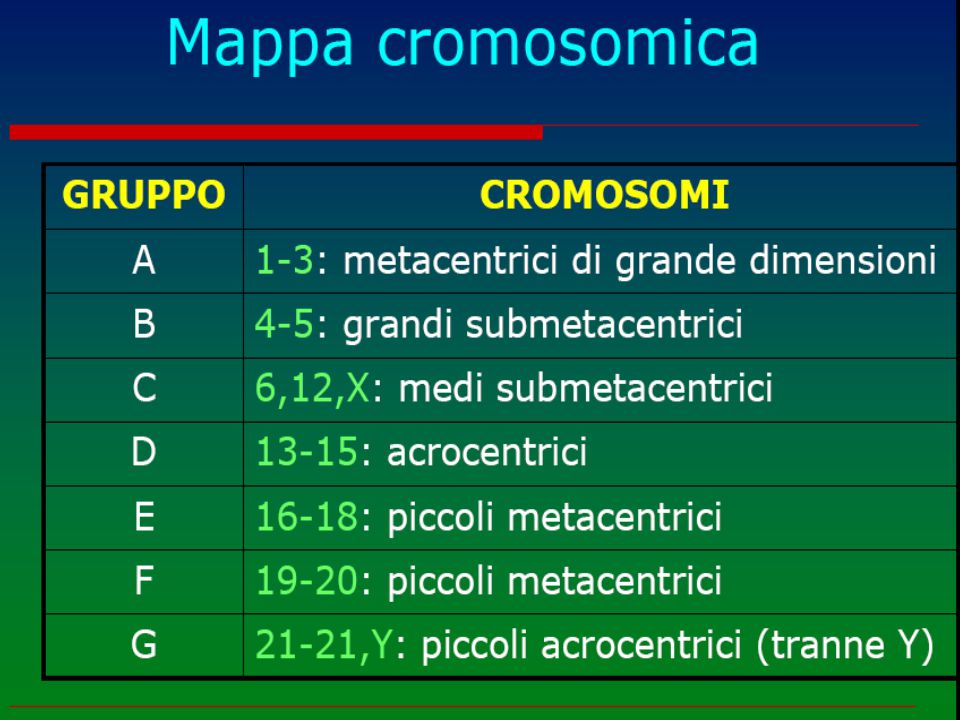
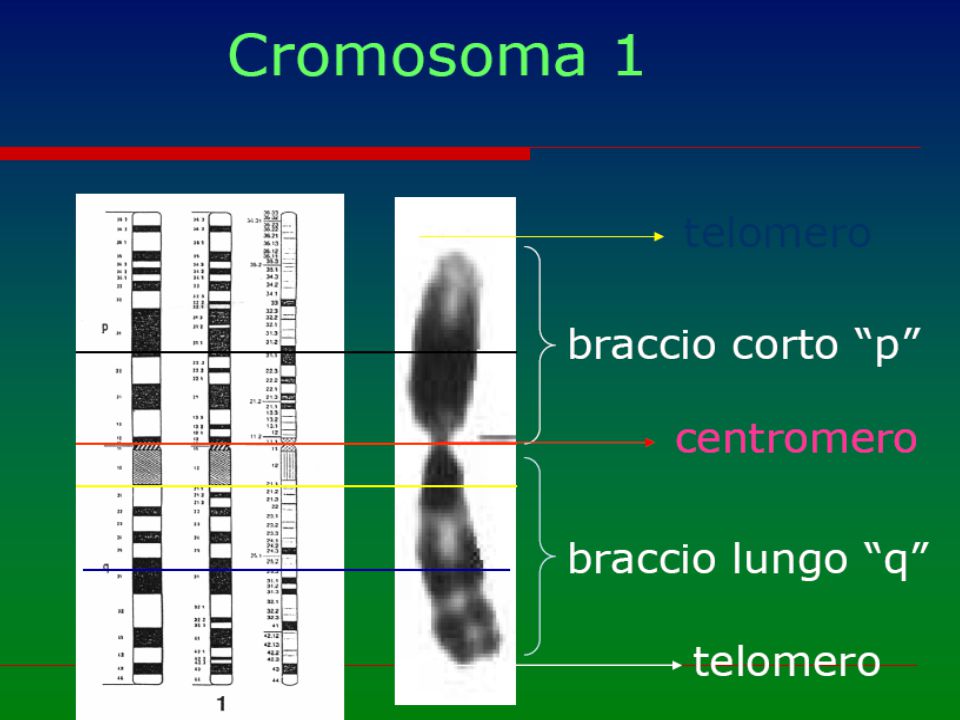
numero anomalo o anomalia strutturale dei cromosomi (“mutazioni genomiche”) Cariotipo Possono essere in linea pura, cioè presenti in tutte le cellule dell'individuo o, più raramente, in mosaico (solo in una percentuale variabile di cellule) ed interessare gli autosomi o i cromosomi sessuali. Ne esistono circa 600 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 X Y
 Cariotipo. Possono essere in linea pura, cioè presenti in tutte le cellule dell individuo o, più raramente, in mosaico (solo in una percentuale variabile di cellule) ed interessare gli autosomi o i cromosomi sessuali. Ne esistono circa X. Y.")
21
Anomalie cromosomiche
Nella grande maggioranza dei casi si tratta di aneuploidie (trisomie, monosomie) la cui causa più comune è una non-disgiunzione della coppia di cromosomi omologhi nella prima divisione meiotica. Circa il 50% degli aborti spontanei mostrano anomalie cromosomiche. La più comunemente osservata nei neonati (1/800) è legata alla presenza di tre cromosomi 21 (sindrome di Down): aumento del rischio con l’età della madre screening prenatale in donne > 35aa Altre anomalie piuttosto frequenti (1/1000) riguardano i cromosomi sessuali: 47XXY (sindrome di klinefelter): alta statura, ipogonadismo, IQ in media ridotto e lievi problemi di apprendimento, possibili problemi comportamentali, infertilità 47XYY alta statura, acne, intelligenza normale, possibili problemi comportamentali, fertili 47XXX “superfemmine”: pubertà precoce, basso IQ e problemi di apprendimento, possibili problemi comportamentali, solitamente fertili 45X (sindrome di Turner): bassa statura, intelligenza normale, infertili, spesso malformazioni aortiche
 la cui causa più comune è una non-disgiunzione della coppia di cromosomi omologhi nella prima divisione meiotica. Circa il 50% degli aborti spontanei mostrano anomalie cromosomiche. La più comunemente osservata nei neonati (1/800) è legata alla presenza di tre cromosomi 21 (sindrome di Down): aumento del rischio con l’età della madre. screening prenatale in donne > 35aa. Altre anomalie piuttosto frequenti (1/1000) riguardano i cromosomi sessuali: 47XXY (sindrome di klinefelter): alta statura, ipogonadismo, IQ in media ridotto e lievi problemi di apprendimento, possibili problemi comportamentali, infertilità. 47XYY alta statura, acne, intelligenza normale, possibili problemi comportamentali, fertili. 47XXX superfemmine : pubertà precoce, basso IQ e problemi di apprendimento, possibili problemi comportamentali, solitamente fertili. 45X (sindrome di Turner): bassa statura, intelligenza normale, infertili, spesso malformazioni aortiche.")
22
Solo una parte di esse sono diagnosticabili in epoca prenatale.
MALATTIE GENICHE Più di 7000 malattie conosciute sono legate ad alterazioni a livello di singoli geni, codificanti la sintesi di proteine strutturali o funzionali anomale. Solo una parte di esse sono diagnosticabili in epoca prenatale. Il loro numero è comunque in continua crescita, in seguito soprattutto al diffondersi delle tecniche di analisi del DNA

23
Un allele può essere DOMINANTE (A) o RECESSIVO (a)
Ciascuno di noi possiede 2 copie di ogni gene contenuto nei cromosomi. Ognuna delle due copia è detta allele. Un allele proviene dal padre (allele paterno) e uno dalla madre (allele materno). Non sempre gli alleli sono uguali fra loro, anzi spesso presentano delle differenze. Gli alleli sono quindi versioni diverse dello stesso gene. Un allele può essere DOMINANTE (A) o RECESSIVO (a) Se un individuo possiede 2 alleli uguali dello stesso gene si dice omozigote Es. AA op. aa Se un individuo possiede 2 alleli diversi dello stesso gene si dice eterozigote Es. Aa
 e uno dalla madre (allele materno). Non sempre gli alleli sono uguali fra loro, anzi spesso presentano delle differenze. Gli alleli sono quindi versioni diverse dello stesso gene. Un allele può essere DOMINANTE (A) o RECESSIVO (a) Se un individuo possiede 2 alleli uguali dello stesso gene si dice omozigote. Es. AA op. aa. Se un individuo possiede 2 alleli diversi dello stesso gene si dice eterozigote. Es. Aa.")
24
Se A = colore giallo dei piselli
e a = colore verde dei piselli AA : si manifesta l’allele dominante giallo Aa : si manifesta l’allele dominante giallo aa : si manifesta l’allele recessivo verde

25
Supponiamo che la mutazione genica sia nell’allele dominante…
aa A a A a aa A a aa Malattia autosomica dominante

26
Supponiamo che la mutazione sia nell’allele recessivo…
aa A A A a A a Malattia autosomica recessiva

27
Malattie monofattoriali
- causate da mutazioni in singoli geni mutazioni puntiformi: missense GAG (glu) > AAG (lys) nonsense CAG (gln) > UAG (stop!) nel promotore alterazione dello splicing del mRNA inserzioni o delezioni puntiformi che alterano la cornice di lettura del DNA delezioni inserzioni duplicazioni espansioni …CAGCAGCAGCAG > …CAGCAGCAGCAG
 > AAG (lys) nonsense CAG (gln) > UAG (stop!) nel promotore. alterazione dello splicing del mRNA. inserzioni o delezioni puntiformi che alterano la cornice di lettura del DNA. delezioni. inserzioni. duplicazioni. espansioni …CAGCAGCAGCAG > …CAGCAGCAGCAG.")
28
Malattie monofattoriali
Singolarmente sono patologie rare, ma nel loro insieme colpiscono circa lo 0.5% di neonati. Si manifestano in prevalenza in età pediatrica (il 5-10% dei bambini ospedalizzati lo sono per una malattia genetica), solo il 10% diventa clinicamente evidente dopo la pubertà. Possiamo distinguere queste malattie in base alla posizione del gene affetto sul cromosoma: autosomiche legate a cromosomi sessuali (X-linked) in base all’espressione fenotipica dominanti (eterozigoti e omozigoti affetti) recessive (solo omozigoti affetti) La diagnosi e l’interpretazione del meccanismo di trasmissione in famiglie è complicata da: eterogeneità: fenotipi simili possono essere causati da difetti diversi dello stesso gene o da mutazioni in geni diversi; penetranza incompleta: il genotipo patologico non si esprime costantemente in fenotipo; espressività: il genotipo patologico si esprime in fenotipi di gravità variabile; variabilità nell’età di comparsa: non tutte le malattie genetiche sono congenite (manifeste clinicamente alla nascita); effetto del sesso: vi sono malattie genetiche autosomiche che si manifestano esclusivamente, o con più alta frequenza, in un determinato sesso; anticipazione e instabilità: tendenza della malattia genetica a manifestarsi in successive generazioni ad una età sempre più precoce e con sintomi sempre più gravi
, solo il 10% diventa clinicamente evidente dopo la pubertà. Possiamo distinguere queste malattie in base alla posizione del gene affetto sul cromosoma: autosomiche. legate a cromosomi sessuali (X-linked) in base all’espressione fenotipica. dominanti (eterozigoti e omozigoti affetti) recessive (solo omozigoti affetti) La diagnosi e l’interpretazione del meccanismo di trasmissione in famiglie è complicata da: eterogeneità: fenotipi simili possono essere causati da difetti diversi dello stesso gene o da mutazioni in geni diversi; penetranza incompleta: il genotipo patologico non si esprime costantemente in fenotipo; espressività: il genotipo patologico si esprime in fenotipi di gravità variabile; variabilità nell’età di comparsa: non tutte le malattie genetiche sono congenite (manifeste clinicamente alla nascita); effetto del sesso: vi sono malattie genetiche autosomiche che si manifestano esclusivamente, o con più alta frequenza, in un determinato sesso; anticipazione e instabilità: tendenza della malattia genetica a manifestarsi in successive generazioni ad una età sempre più precoce e con sintomi sempre più gravi.")
29
Malattie autosomiche dominanti
Rappresentano circa il 50% delle malattie genetiche monofattoriali. Nelle forme familiari, caratteristicamente, ogni figlio/a ha un genitore affetto. Spesso il paziente affetto ha una fitness ridotta, con diminuita capacità di riprodursi. La malattia è quindi mantenuta nella popolazione per la comparsa di mutazioni ex novo (figli affetti con genitori sani). Gli omozigoti sono rarissimi se non per consanguineità e hanno in genere una sintomatologia più severa rispetto all’eterozigote. Neurofibromatosi tipo 1 (1/ eteroziogoti): espressività variabile (macchie caffelatte / tumori fibromatosi della pelle / tumori maligni del SNC) 25-50% dei casi causati da mutazioni ex novo diagnosi è solitamente di tipo clinico talvolta è le diagnosi nel figlio che permette di scoprire la malattia nel genitore (portatore di una variante poco evidente) Possibilità di test genetico per diagnosi prenatale ! Impossibilità di predire la gravità della malattia Corea di Huntington (circa 1/3.000 eterozigoti in Europa occidentale): disturbi neurologici e demenza progressiva con insorgenza in genere dopo i 40aa; nessuna terapia possibile. malattia dovuta a mutazione nel gene IT15 sul cromosoma 4p16.3 (per aumento delle ripetizioni CAG nel primo esone del gene).
. Gli omozigoti sono rarissimi se non per consanguineità e hanno in genere una sintomatologia più severa rispetto all’eterozigote. Neurofibromatosi tipo 1 (1/ eteroziogoti): espressività variabile (macchie caffelatte / tumori fibromatosi della pelle / tumori maligni del SNC) 25-50% dei casi causati da mutazioni ex novo. diagnosi è solitamente di tipo clinico. talvolta è le diagnosi nel figlio che permette di scoprire la malattia nel genitore (portatore di una variante poco evidente) Possibilità di test genetico per diagnosi prenatale. ! Impossibilità di predire la gravità della malattia. Corea di Huntington (circa 1/3.000 eterozigoti in Europa occidentale): disturbi neurologici e demenza progressiva con insorgenza in genere dopo i 40aa; nessuna terapia possibile. malattia dovuta a mutazione nel gene IT15 sul cromosoma 4p16.3 (per aumento delle ripetizioni CAG nel primo esone del gene).")
30
Malattie autosomiche recessive
Rappresentano circa il 30% delle malattie genetiche monofattoriali. I genitori sono solitamente eterozigoti (portatori sani), con il 25% di rischio di generare un figlio omozigote. Classicamente il pedigree mostra solo un membro della famiglia affetto o, al massimo, fratelli affetti. Screening degli eterozigoti Per alcune patologie, la percentuale di portatori sani nella popolazione è estremamente elevata, spesso perché la condizione di eterozigoti dà un vantaggio selettivo: Beta-talassemia: in Sardegna il 12% degli individui è portatore sano; lo screening ha consentito di ridurre ad un terzo il numero di bambini affetti nati ogni anno. La più comune malattia AR in Europa è la fibrosi cistica (1/2000 neonati affetti; circa il 5% degli individui portatori sani). Difetto del trasporto di ioni attraverso la membrana cellulare di ghiandole esocrine (polmoni, pancreas, ghiandole sudoripare) con insufficienza pancreatica ed infezioni polmonari croniche. Eterogeneità con numerose varianti alleliche che determinano il difetto della proteina (in Italia il 45% dei casi sono dovuti ad una specifica mutazione “ΔF508”: perdita di 3 basi con conseguente mancanza di una fenilalanina nella catena aminoacidica della proteina); Conseguente difficoltà ad impostare uno screening degli eterozigoti; Possibilità di screening neonatale.
, con il 25% di rischio di generare un figlio omozigote. Classicamente il pedigree mostra solo un membro della famiglia affetto o, al massimo, fratelli affetti. Screening degli eterozigoti. Per alcune patologie, la percentuale di portatori sani nella popolazione è estremamente elevata, spesso perché la condizione di eterozigoti dà un vantaggio selettivo: Beta-talassemia: in Sardegna il 12% degli individui è portatore sano; lo screening ha consentito di ridurre ad un terzo il numero di bambini affetti nati ogni anno. La più comune malattia AR in Europa è la fibrosi cistica (1/2000 neonati affetti; circa il 5% degli individui portatori sani). Difetto del trasporto di ioni attraverso la membrana cellulare di ghiandole esocrine (polmoni, pancreas, ghiandole sudoripare) con insufficienza pancreatica ed infezioni polmonari croniche. Eterogeneità con numerose varianti alleliche che determinano il difetto della proteina (in Italia il 45% dei casi sono dovuti ad una specifica mutazione ΔF508 : perdita di 3 basi con conseguente mancanza di una fenilalanina nella catena aminoacidica della proteina); Conseguente difficoltà ad impostare uno screening degli eterozigoti; Possibilità di screening neonatale.")
31
MalattieX-linked Sono in grande maggioranza a carattere recessivo:
generalmente ristrette ai maschi, perché le femmine per manifestare la malattia devono essere omozigoti; se un maschio affetto si riproduce con una femmina normale tutti i suoi figli saranno sani e le figlie portatrici; se una donna portatrice si riproduce con un maschio sano, tutti i figli/e avranno il 50% di probabilità di essere affetti. Distrofia muscolare di Duchenne (circa 1/3000 bambini affetti; circa 1/2500 donne portatrici): difetto del gene che sintetizza la distrofina, una proteina che si lega alla membrana delle fibre muscolari e ne preserva l’integrità; progressiva debolezza muscolare (diagnosi clinica tardiva: 2-6aa) con morte entro i 30aa (fitness ridotta, alta percentuale di mutazioni ex novo); gene molto grande (più di due milioni di basi) con numerose varianti alleliche patologiche (70% delezioni) ed espressività variabile genotipo-correlata (in-frame mutations vs. frame-shift mutations); impossibile lo screening degli eterozigoti; possibile diagnosi prenatale in famiglie con variante patologica nota o con figlio affetto (mutazione ex novo vs. familiarità).
: difetto del gene che sintetizza la distrofina, una proteina che si lega alla membrana delle fibre muscolari e ne preserva l’integrità; progressiva debolezza muscolare (diagnosi clinica tardiva: 2-6aa) con morte entro i 30aa (fitness ridotta, alta percentuale di mutazioni ex novo); gene molto grande (più di due milioni di basi) con numerose varianti alleliche patologiche (70% delezioni) ed espressività variabile genotipo-correlata (in-frame mutations vs. frame-shift mutations); impossibile lo screening degli eterozigoti; possibile diagnosi prenatale in famiglie con variante patologica nota o con figlio affetto (mutazione ex novo vs. familiarità).")
32
Malformazioni congenite
Circa il 2% dei neonati è affetto da un difetto morfologico, che può interessare un singolo organo o distretto del corpo o presentarsi in associazione ad altre malformazioni e spesso anche a deficit mentali e motori, configurando talora vere e proprie sindromi. Cause: anomalie cromosomiche difetti genetici esposizione a teratogeni (farmaci, radiazioni, virus o malattie materne come il diabete o l'epilessia,..).
.")
33
Infezioni fetali Causa di anomalie, disfunzioni d'organo,
difetti dell'accrescimento morte in utero. Le principali possono essere anche diagnosticabili in epoca prenatale con differenti modalità (analisi del DNA, indagini immunoematologiche, evidenziazione diretta o in coltura dell'agente patogeno, dimostrazione ecografica di eventuali anomalie fetali) Es: rosolia, toxoplasmosi, infezione da citomegalovirus, varicella, infezione da parvovirus B19.
 Es: rosolia, toxoplasmosi, infezione da citomegalovirus, varicella, infezione da parvovirus B19.")
34
La diagnosi prenatale comprende l'insieme delle procedure che permettono di riconoscere o escludere la presenza nel feto di anomalie congenite

35
Sono spesso diagnosticabili per mezzo dell'ecografia, attraverso un accurato
studio morfologico fetale particolarmente nel secondo trimestre

36
Tecniche di screening prenatale
Lo screening della patologia congenita fetale si avvale oggi di varie tecniche indirette che permettono, attraverso la valutazione nel sangue materno di determinate sostanze di origine fetale o placentare o di caratteristiche strutturali del feto rilevabili con l'ecografia, di individuare situazioni che possono configurare un rischio accresciuto di anomalie fetali, soprattutto cromosomiche.

37
Esse possono essere quindi usate in alternativa o anche in integrazione tra loro, nelle gravidanze considerate a basso rischio, per selezionare i casi a rischio più elevato, da indirizzare alla diagnosi prenatale. Il vantaggio di tali tecniche è naturalmente rappresentato dalla loro mancanza di invasività sul feto,che le rende appunto utilizzabili su larga scala su tutta la popolazione ostetrica, senza rischi di complicazioni sul decorso della gravidanza

38
Il limite è però dato dal fatto di non dare una diagnosi diretta, ma una indicazione di rischio che, in ultima analisi, non risolve l'attesa diagnostica della gravida

39
Doppio, triplo, quadruplo test
TECNICHE NON INVASIVE Doppio, triplo, quadruplo test Test del primo e secondo trimestre basati sulla determinazione dei livelli di alfa- fetoproteina, beta-hCG libera o hCG totale (doppio test) oltre a estriolo non coniugato (triplotest) e inibina A (quadruplo test), associati all'età materna
 oltre a estriolo non coniugato. (triplotest) e inibina A (quadruplo test), associati all età materna.")
40
L'alfafetoproteina è una proteina prodotta dal fegato fetale e dal sacco vitellino utilizzata per lo screening della Sindrome di Down e dei difetti del tubo neurale. In presenza di Sindrome di Down diminuisce di circa il 25-30% nel liquido amniotico e nel sangue materno. In caso di difetti del tubo neurale o della parete addominale, aumenta in maniera significativa, indicando in questo caso l'opportunità di eseguire un esame ecografico per lo studio della morfologia fetale.

41
L‘Estriolo non coniugato è un ormone steroideo, prodotto dal sincizio trofoblasto, che ha precursori prodotti dal fegato fetale e come l'AFP diminuisce di circa il % in caso di Sindrome di Down. L'HCG, la gonadotropina corionica umana è anch'essa un prodotto di secrezione del sincizio trofoblasto; ha circa un valore doppio nelle gravidanze complicate da una Sindrome di Down.

42
TECNICHE NON INVASIVE Test combinato
Test del primo trimestre basato sulla combinazione della misura della translucenza nucale e dei livelli plasmatici di beta-hCG e proteina plasmatica A associata alla gravidanza (PAPP-A) con l'età materna. L'epoca migliore per l'esecuzione, in termini di efficacia, è la 11a settimana.
 con l età materna. L epoca migliore per l esecuzione, in termini di efficacia, è la 11a settimana.")
43
TECNICHE NON INVASIVE Attualmente i markers ecografici ritenuti più suggestivi di anomalie cromosomiche del primo trimestre sono: 1.Translucenza nucale 2.Ossa Nasali

44
(nuchal translucency,NT)
TECNICHE NON INVASIVE Translucenza nucale (nuchal translucency,NT) Si intende lo spessore massima dell’area nucale, compresa fra la cute ed i tessuti molli sovrastanti la colonna vertebrale cervicale, misurabile ecograficamente a settimane complete di gestazione
 Si intende lo spessore massima dell’area nucale, compresa fra la cute ed i tessuti molli sovrastanti la colonna vertebrale cervicale, misurabile ecograficamente a settimane complete di gestazione.")
45
Identificazione ecografica
TECNICHE NON INVASIVE Identificazione ecografica Delle Ossa Nasali I soggetti Down, di solito hanno il naso piccolo poco sviluppato. È stato recentemente dimostrato, che se si effettua un’ecografia tra alla 12a - 14a settimana, nel 98% dei feti Down, le ossa nasali sono assenti, mentre in un feto sano, sono sempre presenti. Associando la valutazione delle ossa nasali ad altri test,come ad esempio la Traslucenza Nucale,si raggiunge una detection rate del 93% con percentuale di falsi positivi del 2% - 3%. Svantaggio: E‘ una metodica ancora poco diffusa che necessita di un ecografo ad alta risoluzione

46
Ecografia morfologia di II livello
TECNICHE NON INVASIVE Ecografia morfologia di II livello L'ecografia morfologica è eseguita per studiare l'anatomia del feto ed escludere circa il 90% delle malformazioni gravi. Tale indagine è condotta con grande accuratezza ed è perciò nota come Ecografia di II livello. Nel corso dell'esame ampio spazio viene dato alla valutazione dell'anatomia e della funzione del cuore fetale (ecocardiografia)
")
47
ECOGRAFIA MORFOLOGICA
Viene eseguita dalla 20 alla 24 settimana di gestazione. La si esegue in questo periodo specifico per due ragioni: Il feto è nelle migliori condizioni per essere studiato, in quanto il rapporto fra le dimensioni del feto e la quantità di liquido amniotico è ottimale 2. Dopo tale epoca la Legge non permette l'interruzione della gravidanza anche se il feto è affetto da gravi malformazioni

48
L'ecografia morfologica a fini puramente conoscitivi può essere eseguita anche più tardivamente ed è comunque utile. Qualora infatti si identificassero patologie malformative potrebbe risultare determinante fare nascere il bambino in strutture particolarmente attrezzate allo scopo. Tale esame dipende in misura quasi totale dall'esperienza e dalla capacità dell'operatore unitamente all'impiego di un ecografo di qualità elevatissima. Nonostante ciò non tutti i quadri patologici sono diagnosticabili in utero; ciò dipende anche dall'ecogenicità della paziente (più aumenta lo spessore del pannicolo adiposo meno chiare saranno le immagini) e dalla quantità di liquido amniotico e dalla posizione fetale.
 e dalla quantità di liquido amniotico e dalla posizione fetale.")
49
TECNICHE INVASIVE Le indicazioni all'esame sono:
età materna avanzata (>35 anni) precedente figlio affetto da anomalia cromosomica genitore portatore di anomalia cromosomica bilanciata familiarità per malattie congenite del metabolismo anomalie strutturali del feto all'esame ecografico di routine test di screening per sindrome di Down positivo motivazioni personali
 precedente figlio affetto da anomalia cromosomica. genitore portatore di anomalia cromosomica. bilanciata. familiarità per malattie congenite del metabolismo. anomalie strutturali del feto all esame ecografico. di routine. test di screening per sindrome di Down positivo. motivazioni personali.")
50
AMNIOCENTESI

51
AMNIOCENTESI Tecnica invasiva di diagnosi prenatale che si esegue nel secondo trimestre di gravidanza mediante l'introduzione per via addominale di un ago in cavità amniotica ecoguidato per il prelievo di ml di liquido. Sul liquido amniotico e sulle cellule fetali prelevate possono essere effettuate indagini citogenetiche per la diagnosi di: anomalie cromosomiche e di sesso per le malattie legate al cromosoma X

52
AMNIOCENTESI Dosaggio dell'alfa-fetoproteina per la diagnosi dei difetti del tubo neurale (attualmente la diagnosi si esegue con l'esame ecografico assieme alla determinazione del livello di alfa-fetoproteina sierica materna a settimane di gestazione) Indagini biochimiche per la diagnosi di errori congeniti del metabolismo
 Indagini biochimiche per la diagnosi di errori congeniti del metabolismo.")
53
VILLOCENTESI

54
TECNICHE INVASIVE Villocentesi
La villocentesi è il prelievo di tessuto placentare Si esegue tra 10 e 12 settimane compiute di gravidanza e permette di diagnosticare le anomalie dei cromosomi del feto (tra cui la trisomia 21), le più gravi e frequenti malattie genetiche malattie genetiche (fibrosi cistica, sindrome dell'X fragile, sordità). Essa consiste nel prelievo, sotto controllo ecografico continuo di alcuni frammenti di tessuto coriale,mediante l'introduzione pervia trans-addominale di un ago
, le più gravi e frequenti malattie genetiche malattie genetiche (fibrosi cistica, sindrome dell X fragile, sordità). Essa consiste nel prelievo, sotto controllo ecografico continuo di alcuni frammenti di tessuto coriale,mediante l introduzione pervia trans-addominale di un ago.")
Presentazioni simili







 Dominanza incompleta>")

 è dominante sul pelo liscio ( r )>")















