Scaricare la presentazione
1
Metodo MCSCF (Multi-Configuration SCF)
")
2
Ci sono problemi che non possono essere descritti a livello di funzione monodeterminantale.
Rottura o formazione di un legame H2 Descrizione degli stati di transizione. Migliore descrizione dello stato fondamentale o degli stati eccitati. Alcune molecole non sono ben descritte da un singolo determinante, per esempio O3.

3
Complessi dei metalli di transizione.
Stati elettronici eccitati. ……

4
Funzione multideterminantale
Espansione con tutte le configurazioni possibili (full CI): qualunque base di orbitali molecolari darebbe lo stesso risultato, cioè il limite dell’insieme di base. Espansione troncata: la scelta degli orbitali diventa importante.
: qualunque base di orbitali molecolari darebbe lo stesso risultato, cioè il limite dell’insieme di base. Espansione troncata: la scelta degli orbitali diventa importante.")
5
Nel metodo CI si ottimizzano i coefficienti CI tenendo fissi gli orbitali molecolari: problema lineare di ottimizzazione dei coefficienti CI. Il miglior risultato per un’espansione CI troncata si ottiene ottimizzando gli orbitali oltre ai coefficienti CI. Questi metodi non sono lineari e si devono quindi risolvere in maniera autoconsistente da cui il nome MCSCF (Multi-Configuration Self Consistent Field)
")
6
Vantaggi possono descrivere correttamente la rottura di un legame possono dare una descrizione compatta della l’espansione è coerente se la lista delle configurazioni è scelta opportunamente sono variazionali Svantaggi ci,r e Cj sono accoppiati: ottimizzazione di molti parametri con molti minimi locali, la convergenza è spesso un problema se le configurazioni sono poche non viene introdotta molta correlazione dinamica.

7
Problema Quali configurazioni inserire nell’espansione ? L’ottimizzazione degli orbitali non può correggere la mancanza di configurazioni importanti nell’espansione. Esempio H2 se introduciamo nell’espansione solo configurazioni che descrivono la correlazione radiale e la correlazione destra-sinistra, l’ottimizzazione degli orbitali non può comunque introdurre la correlazione angolare.

8
Esempio O3 12 + S + D configurazioni c1 = c2 = (12 + 22) + S + D configurazioni c1 = c2 = Il carattere di diradicale dell’ozono dipende da qualche configurazione generata per eccitazione a partire da 22 1 2

9
HF e MCSCF Sia HF che MCSCF ottimizzano gli orbitali molecolari.
HF usa un determinante singolo, mentre MCSCF usa molti determinanti. Metodi avanzati per trattare la correlazione elettronica costruiscono la funzione d’onda basandosi su MO ottimizzati a livello HF — metodi con funzione di riferimento monodeterminantale, esempio CI. MO ottimizzati a livello MCSCF — metodi con funzione di riferimento multideterminantale, esempio MR-CI.

10
Metodo CASSCF [n,m] CASSCF n elettroni in m orbitali
Il metodo CASSCF (Complete Active Space Self Consistent Field) evita il problema della selezione delle configurazioni includendo tutte le configurazioni possibili su un gruppo molto limitato di elettroni e di orbitali. [n,m] CASSCF n elettroni in m orbitali Si ottimizzano sia gli orbitali che i coefficienti dell’espansione. Lo spazio degli orbitali viene diviso in : un gruppo di orbitali inattivi che restano doppiamente occupati. un gruppo di orbitali attivi che vengono occupati in tutti i modi possibili un gruppo di orbitali virtuali che non vengono considerati nel calcolo.
![Metodo CASSCF [n,m] CASSCF n elettroni in m orbitali](http://slideplayer.it/slide/587805/2/images/10/Metodo+CASSCF+%5Bn%2Cm%5D+CASSCF+n+elettroni+in+m+orbitali.jpg "Il metodo CASSCF (Complete Active Space Self Consistent Field) evita il problema della selezione delle configurazioni includendo tutte le configurazioni possibili su un gruppo molto limitato di elettroni e di orbitali. [n,m] CASSCF n elettroni in m orbitali. Si ottimizzano sia gli orbitali che i coefficienti dell’espansione. Lo spazio degli orbitali viene diviso in : un gruppo di orbitali inattivi che restano doppiamente occupati. un gruppo di orbitali attivi che vengono occupati in tutti i modi possibili. un gruppo di orbitali virtuali che non vengono considerati nel calcolo.")
11
Full CI e ottimizzazione degli orbitali in uno spazio attivo piccolo.
[6,6]CASSCF elettroni in 6 orbitali orbitali virtuali orbitali attivi orbitali inattivi 1 2 3 4 5 6 7 8 9 H2O

12
L’ottimizzazione degli orbitali non può compensare l’esclusione di orbitali importanti per la descrizione del processo in esame. Il problema è spostato dalla scelta delle configurazioni alla scelta degli orbitali. In alcuni casi la scelta è immediatamente evidente come nei due seguenti esempi.

13
La scelta dello spazio attivo non è sempre ovvia (Intuizione chimica ed esperienza). La convergenza può essere lenta. In generale, i metodi MCSCF sono utilizzati per ottenere una migliore descrizione delle principali caratteristiche della struttura elettronica prima di tentare il calcolo della maggior parte dell’Energia di correlazione. Si recupera la correlazione statica. La correlazione dinamica è recuperata attraverso un calcolo CI. MR-CI. Le funzioni d’onda CASSCF danno eccellenti stati di riferimento per recuperare una grossa frazione dell’energia di correlazione dinamica. Un calcolo CISD a partire da una funzione di riferimento [n,m]-CASSCF è indicato come Multi-Reference CISD (MR-CISD). Un calcolo MR-CISD può avvicinarsi in accuratezza al calcolo Full CI sulla stessa base, anche se a rigore non gode della proprietà di coerenza.
. Un calcolo MR-CISD può avvicinarsi in accuratezza al calcolo Full CI sulla stessa base, anche se a rigore non gode della proprietà di coerenza.")
14
Funzione d’onda Hartree-Fock
vincoli Funzione d’onda vincolo di equivalenza monodeterminantale stessa parte spaziale Funzioni d’onda plurideterminantali

15
Metodo UHF UHF : Unrestricted Hartree-Fock
Si può scrivere una funzione d’onda in forma monodeterminantale che descriva correttamente la dissociazione togliendo il vincolo di equivalenza, cioè associando ad un elettrone con spin una parte spaziale diversa da quella associata all’elettrone con spin . Per la molecola di idrogeno UHF= det per R può tendere a 1sA e ad 1sB: quindi la dissociazione è descritta correttamente.

16
orbitali UHF per H2

17
La funzione d’onda UHF può essere usata per descrivere la rottura del legame
H2 (2) H(1sA) + H(1sB) H3C-CH3 (2) H3C + CH3 ’
 H(1sA) + H(1sB) H3C-CH3 (2) H3C + CH3. 2 ’")
18
Problema della contaminazione di spin
La funzione UHF è autofunzione di Sz La funzione UHF non è autofunzione di S2 La descrizione dello stato di spin della molecola non è corretta.

19
UHF è una miscela di stati di spin diversi.
Si può valutare il grado di contaminazione calcolando il valore medio di S2. Per uno stato di doppietto S=1/2 e quindi S(S+1)=3/4=0.75. Tanto più grande di 0.75 è il valor medio di S2, tanto più la funzione d’onda è contaminata da componenti di quartetto, sestetto, ...
=3/4=0.75. Tanto più grande di 0.75 è il valor medio di S2, tanto più la funzione d’onda è contaminata da componenti di quartetto, sestetto, ...")
20
Il metodo UHF descrive in forma semplice qualunque sistema con N elettroni e N elettroni , quindi anche sistemi a guscio aperto. Se vogliamo che l’Energia abbia un’espressione semplice occorre imporre l’ortogonalità della base. L’ottimizzazione di una funzione UHF porta alle equazioni di Pople-Nesbet

21
Due equazioni accoppiate attraverso l’interazione Coulombiana.
Nell’approssimazione LCAO si ottiene Si determinano due insiemi di coefficienti diversi per gli orbitali associati ad elettroni con spin e gli orbitali associati ad elettroni con spin .

22
Si può ovviare al fatto che UHF non è autofunzione di S2 introducendo una funzione ROHF (Restricted Open shell Hartree-Fock) in cui gli elettroni e occupano lo stesso orbitale spaziale. UHF ROHF

23
Atomo di Li UHF =1s1s’2s ROHF =1s1s2s Singoletto Doppietto
3 RHF Singoletto ROHF Doppietto UHF Energia

24
Metodo UHF e metodo ROHF
Vantaggi EUHF < EROHF UHF descrive correttamente il processo di dissociazione è più facile da calcolare Svantaggi UHF non è autofunzione di spin (S2)
")
25
Polarizzazione di Spin
Spettro ESR Atomo H 2 linee: la separazione dipende dall’accoppiamento iperfine. L’interazione di contatto di Fermi dipende dalla densità elettronica al nucleo a(nucleo) ge gN |(r=0)|2 e quindi solo dagli orbitali s.
 ge gN |(r=0)|2. e quindi solo dagli orbitali s.")
26
Polarizzazione di spin, ma contaminazione
La polarizzazione di spin è descritta in modo diverso dalle due funzioni UHF e ROHF. Radicale CH L’elettrone nell’orbitale 2p polarizza gli elettroni : l’elettrone vicino ha lo stesso spin, l’altro ha spin opposto. Vicino all’atomo H l’orbitale ≈ orbitale atomico 1s |(r=0)|2 ≠ 0 Polarizzazione di spin, ma contaminazione Manca la polarizzazione di spin Corretta
|2 ≠ 0. Polarizzazione di spin, ma contaminazione. Manca la polarizzazione di spin. Corretta.")
27
Teoria della perturbazione di Rayleigh Schrödinger
Sia dato un problema il cui Hamiltoniano H differisca di poco dall’Hamiltoniano H0 di cui conosciamo le soluzioni. Scriviamo l’Hamiltoniano in termini della perturbazione di H(0). When there is no subscript on the E(0) and Phi(0) symbols this means that we are using the lowest ground state solution of this equation. We also have to assume that we have all the solutions to the unperturbed problem, as these will be used later to provide a complete space in which we can expand the solutions of the perturbed problem H’ è la perturbazione e è un parametro 0 1
. When there is no subscript on the E(0) and Phi(0) symbols this means that we are using the lowest ground state solution of this equation. We also have to assume that we have all the solutions to the unperturbed problem, as these will be used later to provide a complete space in which we can expand the solutions of the perturbed problem. H’ è la perturbazione e è un parametro 0 1.")
28
Sostituendo nell’eq. di Schrödinger ed assumendo che lo stato fondamentale non sia degenere
Indipendenti da l The use of lambda is just a convenient way of switching the perturbation on and off, but also allows us to assign an order the level of perturbation we are going to use. Note at this point we are assuming that the series converges. That each term gets smaller than the previous one. This will happen if the perturbation is small. If we truncate the series (which we will!) then we may no longer have a total energy with a variational bound. E(0) is the zeroth order energy E(1) is the first order correction to the energy E(2) is the second order correction phi(1) is the first order correction to the wavefunction Sostituendo nell’eq. di Schrödinger e raccogliendo i termini con la stessa potenza di , si ottiene un insieme di equazioni.
 then we may no longer have a total energy with a variational bound. E(0) is the zeroth order energy. E(1) is the first order correction to the energy. E(2) is the second order correction. phi(1) is the first order correction to the wavefunction. Sostituendo nell’eq. di Schrödinger e raccogliendo i termini con la stessa potenza di , si ottiene un insieme di equazioni.")
29
Moltiplichiamo ciascuna equazione per F(0) ed integriamo
Assumiamo: For each power of lambda there is an equation that must hold. The level of perturbation theory being used is referred to by the order of the perturbation parameter lambda. Zeroth First Second and Third order expressions. The choice of ‘intermediate normalisation’ makes life simpler later on. To obtain expressions for the energy at each order we multiply each expression through by the unperturbed ground state wavefunction and integrate over all the coordinates. Moltiplichiamo ciascuna equazione per F(0) ed integriamo
 ed integriamo.")
30
Perturbazione all’ordine zero
The main point is that this is only true when phi(0) is the solution of the unperturbed problem. So the zeroth order solution is the solution to the unperturbed problem. But it is worth while working through as it makes you work through the calculation of the hamiltonian expectation value and use the normalisation of the wavefunction.
 is the solution of the unperturbed problem. So the zeroth order solution is the solution to the unperturbed problem. But it is worth while working through as it makes you work through the calculation of the hamiltonian expectation value and use the normalisation of the wavefunction.")
31
Perturbazione al primo ordine
The tricks to remember are that phi(0) and phi(1) or orthogonal. (intermediate normalisation ) Also that the ‘turnover rule’ H12 = (H21)* since we are dealing with real quantities H12 = H21. Finally E(1) is just the expectation value of the perturbation over the zeroth order ground state wavefunction.
 and phi(1) or orthogonal. (intermediate normalisation ) Also that the ‘turnover rule’ H12 = (H21)* since we are dealing with real quantities H12 = H21. Finally E(1) is just the expectation value of the perturbation over the zeroth order ground state wavefunction.")
32
Funzione d’onda al primo ordine
We use the fact that the solutions of the unperturbed Hamiltonian form a complete set of functions, so we can expand our first order wavefunction as a sum over the unperturbed wavefunctions. To keep things clear, the 0 subscript has been included in the ground state unperturbed wavefunction. Note the use of the chosen normalisation. The 1st and higher order wavefunctions has no contribution from the zeroth order, unperturbed wavefunction. So we can replace the sum over all states by a sum over all states except the lowest. To determine the coefficients in the sum we multiply the left of the equations by an excited, unperturbed wavefunction phi(j) and integrate over all space. Orthonormality of the unperturbed states means that this projects out all but that state from the sum. Finally giving an expression for the jth coefficient of the unperturbed wavefunction in 1st order perturbed wavefunction. Each coefficient is the matrix element of the perturbation coupling the ground state unperturbed wavefunction with the excited unperturbed wfn, divided by an energy denominator. Expect the coefficients to get smaller as the states of the unperturbed solution get higher in energy.
 and integrate over all space. Orthonormality of the unperturbed states means that this projects out all but that state from the sum. Finally giving an expression for the jth coefficient of the unperturbed wavefunction in 1st order perturbed wavefunction. Each coefficient is the matrix element of the perturbation coupling the ground state unperturbed wavefunction with the excited unperturbed wfn, divided by an energy denominator. Expect the coefficients to get smaller as the states of the unperturbed solution get higher in energy.")
33
Perturbazione al secondo ordine
The second order energy is a sum over states, other than the unperturbed ground state. Only the zeroth, unperturbed solution is needed. The denominator is the energy gap. The numerator is the square of the matrix element of the perturbation connecting the ground state, unperturbed function with the excited state wavefunctions. One expects that higher energy states will have less contribution to the 2nd order energy. There is a relationship between the perturbation expression and the CI hamiltonian. If the unperturbed hamiltonian is taken to be that which gives the hartree Fock solution, the only matrix elements coupling with the HF wfn are double excitations. If we take the full CI hamiltonian and subtract the hartree fock energy from the digaonal terms, diagonalisation will give the complete correction to the energy. The first order energy is zero <0|H00-H00|0> The second order energy is given by the expressions above and only involves double excitations. E(2) : contributo negativo all’energia
 : contributo negativo all’energia.")
34
Cose da ricordare sulla teoria della perturbazione
La serie può non convergere. La convergenza può essere lenta. Il parametro d’ordine è sempre posto = 1 quando si calcola l’energia finale. L’energia non è un valore superiore rispetto all’energia esatta, non è variazionale. La degenerazione dello stato fondamentale causa problemi. La scelta dell’Hamiltoniano di ordine zero è arbitraria.

35
MBPT o metodi MPn MBPT Many Body Perturbation Theory
MPn teoria della perturbazione Möller-Plesset all’ordine n H = H(0) + H’ Se come Hamiltoniano di ordine zero viene preso l’Hamiltoniano Hartree-Fock si ha il metodo Möller-Plesset.
 + H’ Se come Hamiltoniano di ordine zero viene preso l’Hamiltoniano Hartree-Fock si ha il metodo Möller-Plesset.")
37
L’effetto della perturbazione può essere valutato all’ordine n: MPn
Il primo contributo all’energia di correlazione è dato da MP2

38
Teoria della perturbazione MP2 Möller-Plesset al secondo ordine
Note that starting with the standard expression for the 2nd order correction to the energy, we require the excited states of the zeroth order problem. We can right these down as we did previously, but this time we omit single and triple and higher excitations, as we know these have no contribution (see previous slide) SO we replace the sum of I, by a sum over ij->ab The second order energy is particularly simple, requiring only molecular 2 electron integrals<ij||ab> and the orbital energies.
 SO we replace the sum of I, by a sum over ij->ab. The second order energy is particularly simple, requiring only molecular 2 electron integrals<ij||ab> and the orbital energies.")
39
Convergenza della serie MPn
Problema : la funzione d’onda HF è una buona funzione di ordine zero solo in prossimità della geometria di equilibrio la convergenza dell’espansione si degrada quando la geometria si allontana da quella di equilibrio. Convergenza della serie MPn H2O Eesatta = hartree Eesatta = hartree R = Re R = 2 Re

40
MPn: Curve ad alto ordine
H2 MPn diverge quando il legame è stirato

41
Vantaggi della teoria MPn
Calcoli MP2 su sistemi di dimensioni moderate (~150 funzioni di base) hanno ~ lo stesso costo computazionale di HF. Scalano come M5, ma in pratica molto meno. Godono della proprietà estensiva. Ricuperano ~80-90% dell’energia di correlazione. Estensione al quarto ordine: MP4(SDQ) e MP4(SDTQ). MP4(SDTQ) ricupera ~95-98% dell’energia di correlazione, ma scala come M7. 1) il costo computazionale è decisamente inferiore a CISD 2) MPn gode della proprietà estensiva MP2 è un buon metodo per introdurre la correlazione elettronica.
 hanno ~ lo stesso costo computazionale di HF. Scalano come M5, ma in pratica molto meno. Godono della proprietà estensiva. Ricuperano ~80-90% dell’energia di correlazione. Estensione al quarto ordine: MP4(SDQ) e MP4(SDTQ). MP4(SDTQ) ricupera ~95-98% dell’energia di correlazione, ma scala come M7. 1) il costo computazionale è decisamente inferiore a CISD. 2) MPn gode della proprietà estensiva. MP2 è un buon metodo per introdurre la correlazione elettronica.")
42
Variante della teoria MP: CASPT2
Teoria della perturbazione con funzione di riferimento multi determinantale per lo stato fondamentale e stati eccitati. Espansione CAS. Correzione MP2 per tutte le funzioni di riferimento.

43
Il problema della funzione di riferimento
N2 R

44
Che metodo usare? Quando HF non fornisce una funzione d’onda di ordine zero qualitativamente corretta, usare MCSCF. L’espansione CI troncata non è coerente, non dovrebbe essere usata se altri metodi sono utilizzabili. Gli stati eccitati sono difficili da trattare con i metodi MPn e CC, la scelta è MCSCF e CI (meglio MR-CI). CCSD(T) con grandi insiemi di base può raggiungere l’accuratezza chimica (~ 1 kcal/mole). HF MP2 CISD, CCSD MP4 CCSD(T) Accuratezza e costo computazionale
. CCSD(T) con grandi insiemi di base può raggiungere l’accuratezza chimica (~ 1 kcal/mole). HF. MP2. CISD, CCSD. MP4. CCSD(T) Accuratezza e costo computazionale.")
45
Metodi compositi Due vie per formulare metodi di elevata accuratezza, usando additività estrapolazione

46
Schemi di additività e metodi Gn
Aumenta la dimensione della base DZ TZ funzioni Migliora la correlazione MP2 CCSD(T) Risposta esatta full CI
 Risposta. esatta. full CI.")
47
Teorie Gn Pople: metodo G1 (protocollo empirico) per calcolare un valore dell’energia, equivalente a quello ottenibile attraverso un calcolo (non realizzabile) con base molto grande ed inclusione di una sostanziale percentuale della correlazione, mediante la somma di contributi considerati additivi e determinabili mediante calcoli di dimensioni accettabili. 1 Geometria ottimizzata MP2/6-31G(d) 2 Energia di riferimento MP4/6-311G(d,p) 3 + Correzione per funzioni diffuse MP4/6-311G+(d,p) 4 2df Correzione per ulteriori funzioni di polarizzazione MP4/6-311G(2df,p) 5 +QCI Correzione per correlazione residua QCISD(T)/6-311G(d,p) 6 Correzione empirica n n 7 Energia di punto zero La correzione empirica scelta per ottenere i valori esatti dell’energia di H e H2. Nella versione G2 si aggiungono 2 ulteriori correzioni (+2df) - + - (2df) correzione per non addidività dei contributi delle funzioni diffuse e delle funzioni di polarizzazione (3df,2p) - (2df,p) correzione per ulteriori funzioni di polarizzazione Attualmente esiste una versione G3
 per calcolare un valore dell’energia, equivalente a quello ottenibile attraverso un calcolo (non realizzabile) con base molto grande ed inclusione di una sostanziale percentuale della correlazione, mediante la somma di contributi considerati additivi e determinabili mediante calcoli di dimensioni accettabili. 1. Geometria ottimizzata. MP2/6-31G(d) 2. Energia di riferimento. MP4/6-311G(d,p) 3. + Correzione per funzioni diffuse. MP4/6-311G+(d,p) 4. 2df Correzione per ulteriori funzioni di polarizzazione. MP4/6-311G(2df,p) 5. +QCI Correzione per correlazione residua. QCISD(T)/6-311G(d,p) 6. Correzione empirica n n 7. Energia di punto zero. La correzione empirica scelta per ottenere i valori esatti dell’energia di H e H2. Nella versione G2 si aggiungono 2 ulteriori correzioni. (+2df) - + - (2df) correzione per non addidività dei contributi delle funzioni diffuse e delle funzioni di polarizzazione. (3df,2p) - (2df,p) correzione per ulteriori funzioni di polarizzazione. Attualmente esiste una versione G3.")
48
Deviazione media assoluta su 148 entalpie di formazione per una serie di molecole
G2 ± 1.56 kcal/mole G3 ± 0.94 kcal/mole Deviazione media assoluta complessiva su 299 energie G2 ± 1.48 kcal/mole G3 ± 1.02 kcal/mole

49
Metodi di estrapolazione: metodi CBS
CBS Complete Basis Set Aumenta la dimensione della base DZ E TZ QZ Limite della base

50
Tempi relativi di calcolo con diversi metodi
Metodo Tempo SCF 1 MP2 1.5 MP3 3.6 MP4-SDQ 5.8 CI-SD 17

51
Costo computazionale di HF e di metodi correlati
(SGI R8000 con Gaussian 94) 50 100 150 200 250 300 350 400 C2H6 C3H8 C4H10 C5H12 C6H14 C7H16 C8H18 CCSD/6-31G** Energia CISD/6-31G** Energia MP4/6-31G** Energia DFT BLYP/6-31G* Energia MP2/6-31G** Energia HF/6-31G* Frequenze HF/6-31G* Ottimizzazione Formula Molecolare Tempo in Minuti
 C2H6. C3H8. C4H10. C5H12. C6H14. C7H16. C8H18. CCSD/6-31G** Energia. CISD/6-31G** Energia. MP4/6-31G** Energia. DFT BLYP/6-31G* Energia. MP2/6-31G** Energia. HF/6-31G* Frequenze. HF/6-31G* Ottimizzazione. Formula Molecolare. Tempo in Minuti.")
52
Metodi utilizzati


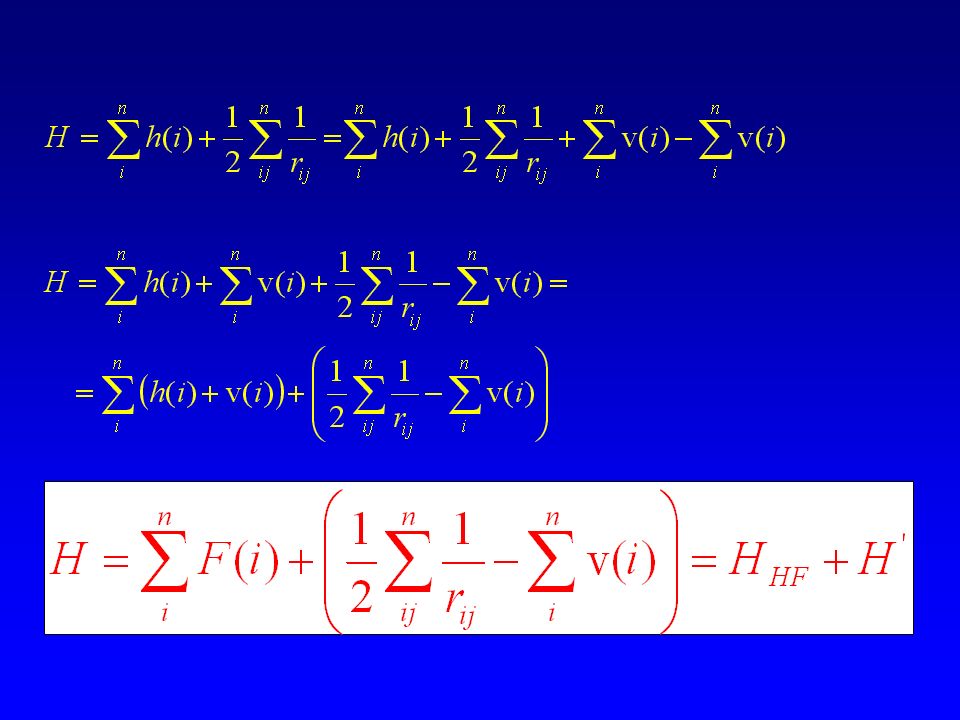


 Neutroni (n°) Elettroni (e) Gli atomi contengono diversi tipi di particelle subatomiche.>")
















