Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Neoplasie Endocrine Multiple (MEN)
Emanuele Bosi Università Vita-Salute San Raffaele A.A. 2009/10

2
Multiple Endocrine Neoplasia (MEN)
Le Neoplasie Endocrine Multiple (Multiple Endocrine Neoplasia, MEN) sono patologie familiari connotate dalla presenza nello stesso paziente di lesioni iperplastiche, adenomatose o adenocarcinomatose in due o più ghiandole endocrine.
 sono patologie familiari connotate dalla presenza nello stesso paziente di lesioni iperplastiche, adenomatose o adenocarcinomatose in due o più ghiandole endocrine.")
3
MEN: general features The MEN syndromes differ from other hereditary cancer syndromes in that most tumor growth occurs in hormone-secreting glands. This feature has two primary consequences of clinical importance: the excess hormone production often results in well-defined hormonal syndromes with characteristic symptoms and medical sequelae. the excess hormone production serves as a sensitive tumor marker that is useful for making a diagnosis, determining response to therapy, and screening asymptomatic patients.

4
Classificazione In base alle ghiandole endocrine interessate si distinguono le seguenti forme: MEN MEN2 Altre (Sindromi miste) Classificazione messa a punto in questi ultimi anni (proposta da Capella, Solcia et al. nel 1995 e adottata dalla Organizzazione Mondiale della Sanità nel 2000) che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.
 che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.")
5
Classificazione: MEN1 MEN 1 - Wermer’s syndrome
Paratiroidi: Iperplasia o adenoma (paratormone) Pancreas endocrino e duodeno: Iperplasia, adenoma o carcinoma (gastrina, insulina, glucagone, somatostatina, PP, VIP) Ipofisi: Iperplasia o adenomi (prolattina, GH, ACTH) Altre manifestazioni cliniche meno comuni: carcinoide, feocromocitoma, lipomi sottocutanei o viscerali Classificazione messa a punto in questi ultimi anni (proposta da Capella, Solcia et al. nel 1995 e adottata dalla Organizzazione Mondiale della Sanità nel 2000) che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.
 Pancreas endocrino e duodeno: Iperplasia, adenoma o carcinoma (gastrina, insulina, glucagone, somatostatina, PP, VIP) Ipofisi: Iperplasia o adenomi (prolattina, GH, ACTH) Altre manifestazioni cliniche meno comuni: carcinoide, feocromocitoma, lipomi sottocutanei o viscerali. Classificazione messa a punto in questi ultimi anni (proposta da Capella, Solcia et al. nel 1995 e adottata dalla Organizzazione Mondiale della Sanità nel 2000) che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.")
6
Classificazione: MEN2 MEN 2A MTC: Carcinoma Midollare della Tiroide
Feocromocitoma Iperplasia o adenoma delle paratiroidi In associazione a: amiloidosi e lichen cutaneo malattia di Hirschsprung FMTC: Carcinoma Midollare Familiare della Tiroide MEN 2B MTC: Carcinoma Midollare della Tiroide Feocromocitoma Neurinomi delle mucose e gastrointestinali Habitus marfanoide Classificazione messa a punto in questi ultimi anni (proposta da Capella, Solcia et al. nel 1995 e adottata dalla Organizzazione Mondiale della Sanità nel 2000) che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.
 che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.")
7
Classificazione: altre, forme miste
Carcinoma midollare familiare della tiroide: almeno 4 membri affetti senza altre endocrinopatie Von Hippel Lindau: feocromocitoma, emangioblastoma retinico o SNC, carcinoma cellule chiare del rene, tumori isole pancreatiche, .. Neurofibromatosi associate a MEN: feocromocitoma, macchie caffè-latte, neurofibromi, .. Sindrome di Cowden: carcinoma non midollare della tiroide (papillare o follicolare), neoplasie di cute, mammella, mucosa orale, utero Carney complex: tumori endocrini (tiroide, ipofisi, corticosurrene), pigmentazione cutanea, mixomi, schwannomi
, neoplasie di cute, mammella, mucosa orale, utero. Carney complex: tumori endocrini (tiroide, ipofisi, corticosurrene), pigmentazione cutanea, mixomi, schwannomi.")
8
Classificazione WHO A revised clinicopathological classification of neuroendocrine tumors of the gastroenteropancreatic tract has been developed under the auspices of the World Health Organization (WHO) according to advances in the field of tumor biology. Solcia E, Kloppel G, Sobin LH (2000) Histological Typing of Endocrine Tumours. World Health Organization International Histological Classification of Tumours. Classificazione messa a punto in questi ultimi anni (proposta da Capella, Solcia et al. nel 1995 e adottata dalla Organizzazione Mondiale della Sanità nel 2000) che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.
 according to advances in the field of tumor biology. Solcia E, Kloppel G, Sobin LH (2000) Histological Typing of Endocrine Tumours. World Health Organization International. Histological Classification of Tumours. Classificazione messa a punto in questi ultimi anni (proposta da Capella, Solcia et al. nel 1995 e adottata dalla Organizzazione Mondiale della Sanità nel 2000) che tiene conto dei dati clinico-patologici e dei parametri morfologici a significato prognostico.")
9
Classificazione WHO: Novità principali
Nomenclatura Abbandono del termine “carcinoide” a favore di tumore o carcinoma: “instead of carcinoid, the WHO classification published in 2000 uses the general terms neuroendocrine tumor and neuroendocrine carcinoma” Utilizzo combinato di dati anatomo-clinici e funzionali Volume, presenza di metastasi, presenza di angioinvasione, tipo di secrezione ormonale, presenza o meno di sindrome clinica associata: “On the basis of localization and of various morphological and biological criteria, we distinguish between benign neuroendocrine tumors, tumors with uncertain malignant potential, and tumors showing low-grade and high-grade malignancy”. Suddivisione per sede Stomaco, pancreas, duodeno, digiuno-ileo, appendice, colon-retto. Novità apportate dalla classificazione WHO: Sforzo di classificazione terminologica: il vecchio termine carcinoide non è adeguato e non viene più utilizzato; Sforzo di unificazione dei parametri utili al clinico: sede, TNM, criteri biologici biologia, funzione del tumore e clinica. La classificazione WHO, quindi, oltre che apportare novità di nomenclatura, richiede una descrizione del tumore che si basi, oltre che sulla sede di origine e sui classici parametri anatomoclinici (dimensioni, presenza di metastasi accertate e di angioinvasione TNM), anche sui parametri biologici della crescita tumorale e sui parametri funzionali, come la secrezione ormonale e l’eventuale presenza di sindrome clinica associata.
, anche sui parametri biologici della crescita tumorale e sui parametri funzionali, come la secrezione ormonale e l’eventuale presenza di sindrome clinica associata.")
10
Classificazione WHO: Criteri
Parametri patologici Clinica Contesto clinico generale Secrezione ormonale Parametri patologici (sede, dimensione, coinvolgimento delle tonache di parete/diffusione extraorgano, indice proliferativo, angio-invasione, linfonodi, metastasi, residuo di malattia): Tumori endocrini ben differenziati (benigni/comportamento biologico incerto) funzionanti e non funzionanti Carcinomi endocrini ben differenziati (basso grado di malignità) funzionanti e non funzionanti Carcinomi endocrini scarsamente differenziati (alto grado di malignità) Carcinomi misti endocrini/esocrini Clinica: Funzionanti/non funzionanti Contesto clinico generale: (stomaco: tumori endocrini associati o meno ad ipergastrinemia) Produzione ormonale: Dimostrabile con metodiche immunoistochimiche
: Tumori endocrini ben differenziati (benigni/comportamento biologico incerto) funzionanti e non funzionanti. Carcinomi endocrini ben differenziati (basso grado di malignità) funzionanti e non funzionanti. Carcinomi endocrini scarsamente differenziati (alto grado di malignità) Carcinomi misti endocrini/esocrini. Clinica: Funzionanti/non funzionanti. Contesto clinico generale: (stomaco: tumori endocrini associati o meno ad ipergastrinemia) Produzione ormonale: Dimostrabile con metodiche immunoistochimiche.")
11
Classificazione WHO: Criteri
Parametri patologici (sede, dimensione, coinvolgimento delle tonache di parete/diffusione extraorgano, indice proliferativo, angio-invasione, linfonodi, metastasi, residuo di malattia): Tumori endocrini ben differenziati (benigni/comportamento biologico incerto) funzionanti e non funzionanti Carcinomi endocrini ben differenziati (basso grado di malignità) funzionanti e non funzionanti Carcinomi endocrini scarsamente differenziati (alto grado di malignità) Carcinomi misti endocrini/esocrini Clinica: Funzionanti/non funzionanti Contesto clinico generale: (stomaco: tumori endocrini associati o meno ad ipergastrinemia) Produzione ormonale: Dimostrabile con metodiche immunoistochimiche
: Tumori endocrini ben differenziati (benigni/comportamento biologico incerto) funzionanti e non funzionanti. Carcinomi endocrini ben differenziati (basso grado di malignità) funzionanti e non funzionanti. Carcinomi endocrini scarsamente differenziati (alto grado di malignità) Carcinomi misti endocrini/esocrini. Clinica: Funzionanti/non funzionanti. Contesto clinico generale: (stomaco: tumori endocrini associati o meno ad ipergastrinemia) Produzione ormonale: Dimostrabile con metodiche immunoistochimiche.")
12
Classificazione WHO Tumori Endocrini ben differenziati a comportamento benigno vs incerto dimensione (1 cm [2 cm per pancreas e appendice]) e angioinvasione Tumori differenziati Benigni A comportamento biologico incerto Tumori Endocrini vs carcinomi infiltrazione della tonaca muscolare (mesenteriolo per appendice) e presenza di metastasi Carcinomi Ben differenziati (basso grado di malignità) Scarsamente differenziati (alto grado di malignità) TE benigni vs Comportamento Incerto dimensione (1 cm, 2 cm per pancreas ed appendice) ed angioinvasione. TE vs CA infiltrazione della tonaca muscolare (mesenteriolo per appendice) e metastasi. Tumori misti Esocrini-endocrini
 e angioinvasione. Tumori differenziati. Benigni. A comportamento biologico incerto. Tumori Endocrini vs carcinomi. infiltrazione della tonaca muscolare (mesenteriolo per appendice) e presenza di metastasi. Carcinomi. Ben differenziati (basso grado di malignità) Scarsamente differenziati (alto grado di malignità) TE benigni vs Comportamento Incerto dimensione (1 cm, 2 cm per pancreas ed appendice) ed angioinvasione. TE vs CA infiltrazione della tonaca muscolare (mesenteriolo per appendice) e metastasi. Tumori misti. Esocrini-endocrini.")
13
MULTIPLE ENDOCRINE NEOPLASIA
MEN TYPE 1 Wermer’s syndrome

14
MEN TYPE 1 Genetic Wermer’s syndrome
Autosomal-dominant condition that occurs as a result of inactivating mutations of MEN1 gene The MEN 1 gene is located at chromosome 11q13 and consist of 10 exons with a 1830-bp coding region that encodes a novel 610-amino acid protein, referred to as MENIN. The presumed unifying mechanism for tumor formation in Men 1 involves loss of MENIN function in a tumor precursor cell.

16
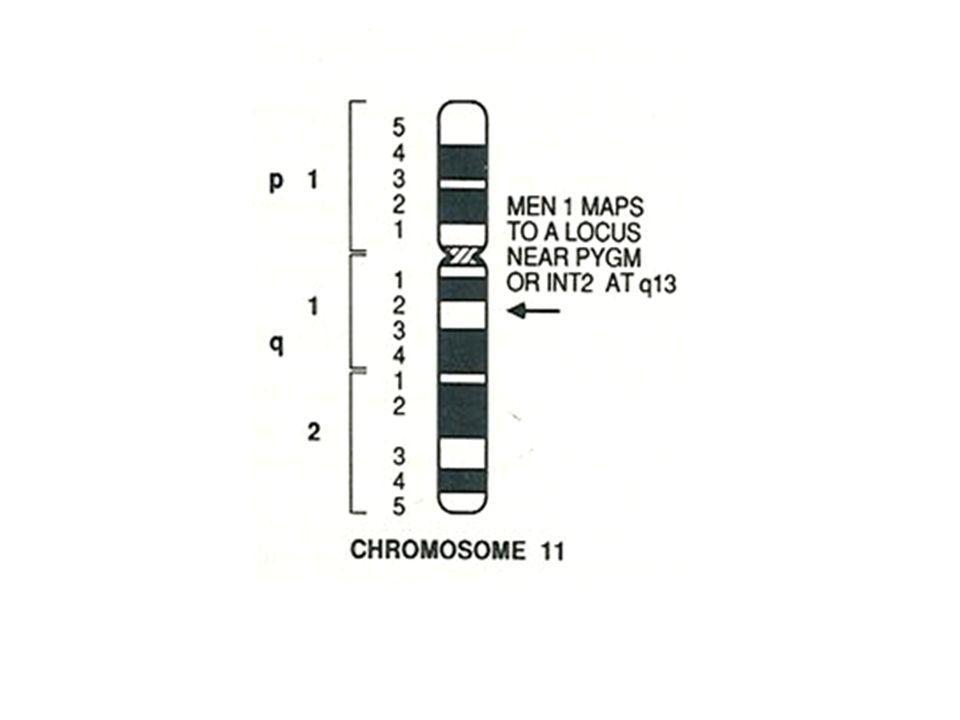
Diagnosi Genetica sostituzione C-G in posizione 1561 dell’esone 10 [sostituzione AA Arg-Gly in posizione 521] sostituzione T-C in posizione 7257 dell’esone 9 [sostituzione AA Phe-Ser in posizione 416] MEN 1 11q13 Nuove mutazioni del gene MEN1
![Diagnosi Genetica sostituzione C-G in posizione 1561 dell’esone 10 [sostituzione AA Arg-Gly in posizione 521]](http://slideplayer.it/slide/984095/3/images/16/Diagnosi+Genetica+sostituzione+C-G+in+posizione+1561+dell%E2%80%99esone+10+%5Bsostituzione+AA+Arg-Gly+in+posizione+521%5D.jpg "sostituzione T-C in posizione 7257 dell’esone 9 [sostituzione AA Phe-Ser in posizione 416] MEN 1 11q13. Nuove mutazioni del gene MEN1.")
17
MEN1: General features Multifocal nature of the disease process within a single organ. Hyperplasia adenoma carcinoma a neoplastic process in one organ may affect the progression in another organ (i.e. pancreatic tumor may stimulate growth of a pituitary tumor). the syndrome evolves in years and the manifestation will in large parte on when the syndrome is identified. The prevalence of MEN1 has been estimated at 1 in 20-40,000 individuals
. the syndrome evolves in years and the manifestation will in large parte on when the syndrome is identified. The prevalence of MEN1 has been estimated at 1 in 20-40,000 individuals.")
18
Percent of MEN 1 Clinical Features Entero-Pancreatic Tumor
Hyperparathyroidism Entero-Pancreatic Tumor Pituitary Adenoma 90-100% 80% 50-60%

19
MEN1: sindromi cliniche da iperplasia, adenomi, carcinomi endocrini
Paratiroidi: Iperparatiroidismo primitivo Pancreas endocrino e duodeno: Gastrinoma (Zollinger-Ellison) Insulinoma Glucagonoma Somatostatinoma VIPoma (Watery Diarrhea Syndrome) PPoma, non secernenti Ipofisi: prolattina, GH, ACTH, non secernenti
 Insulinoma. Glucagonoma. Somatostatinoma. VIPoma (Watery Diarrhea Syndrome) PPoma, non secernenti. Ipofisi: prolattina, GH, ACTH, non secernenti.")
20
Insulinoma: Clinica Neuroglicopenia Eccesso di catecolamine
Cambiamenti di personalità Confusione Epilessia Coma Altri sintomi Appetito Fatica Nausea, vomito Neuropatia periferica Eccesso di catecolamine Diaforesi Pallore Tachicardia

21
Distribuzione del tumore
Glucagonoma: Clinica Segni e sintomi Distribuzione del tumore Diabete Mellito Sindrome endocrina multipla Eritema necrolitico migrante Sindrome neoplastica Sindrome neoplastica Calo ponderale Diarrea Trombosi venosa profonda Anemia Eritema necrolitico migrante 51% 14% 22% Eritema necrolitico migrante: rush localizzato prevalentemente intorno agli orifizi (periorale e perineale), in corrispondenza delle pieghe e delle dita; presenza di chiazze eritematose di colore dal rosa al rosso a margini irregolari e limiti sfumati sormontato da papule e in parte da elementi vescicolo-bollose che evolvono in croste con possibili esiti pigmentari ad andamento centrifugo glucocorticoidi topici associati a permanganato di potassio. 80% MALIGNI
, in corrispondenza delle pieghe e delle dita; presenza di chiazze eritematose di colore dal rosa al rosso a margini irregolari e limiti sfumati sormontato da papule e in parte da elementi vescicolo-bollose che evolvono in croste con possibili esiti pigmentari ad andamento centrifugo glucocorticoidi topici associati a permanganato di potassio. 80% MALIGNI.")
22
Distribuzione del tumore
Gastrinoma: Clinica Segni e sintomi Distribuzione del tumore Ulcere Sintomi da reflusso Dolore addominale Diarrea 5% Il gastrinoma origina dalle cellule G, localizzate prevalentemente nel duodeno prossimale (83%) e in minor misura nell’antro gastrico, e da popolazioni cellulari denominate D1 a sede pancreatica; l’espressione genica della gastrina nel pancreas dei mammiferi è limitata alla vita fetale, per cui i gastrinomi pancreatici sono considerati tumori “ectopici”, mentre i gastrinomi del duodeno, del digiuno e dello stomaco, dove le cellule G sono normalmente presenti, sono considerati tumori “eutopici”. I gastrinomi rappresentano il tumore neuroendocrino digestivo più frequente nell’ambito della MEN 1: circa il 40% dei casi MEN 1 presenta il gastrinoma e circa il 25% dei casi totali di gastrinoma è associato a MEN 1. Il tumore produce gastrina, un ormone peptidico in grado di stimolare la secrezione acida dello stomaco. Tumori con espressione immunoistochimica di gastrina, senza evidenza clinica di ZES non dovrebbero essere considerati gastrinomi. La sede di più frequente insorgenza è il pancreas, e l’incidenza calcolata è di circa 1/ persone/anno. Sono possibili comunque altre localizzazioni, digestive o addirittura extradigestive. Triangolo del gastrinoma: testa pancreatica-porzione superiore e discendente del duodeno-linfonodi. Il duodeno rappresenta la localizzazione principale dei gastrinomi associati a MEN 1, che sono spesso di piccole dimensioni (<1 cm) e multifocali. Altri siti primari di insorgenza del gastrinoma sono lo stomaco, il digiuno, le vie biliari, fegato, reni, mesenterio, cuore e ovaio; l’ipotesi di insorgenza primaria linfonodale dei gastrinomi è controversa: i gastrinomi duodenali in particolare, infatti, possono metastatizzare quando ancora molto piccoli, suggerendo che i cosiddetti gastrinomi linfonodali siano in realtà metastasi di microgastrinomi duodenali occulti. La malignità riguarda circa il 60% dei casi. Il dolore addominale (78%) e la diarrea (40%) sono le manifestazioni cliniche più importanti. Il dolore addominale è da riferire alla malattia ulcerosa, all’iperperistaltismo e all’infiltrazione neoplastica di rami nervosi. La lesione peptica si localizza nel 75% dei casi nella porzione prossimale del duodeno e meno comunemente nello stomaco distale. La diarrea e la steatorrea sono causate principalmente alla ridotta capacità di assorbimento intestinale per aumentata secrezione gastrica ed all’inattivazione delle lipasi pancreatiche e alla precipitazione de gli acidi biliari con conseguente malassorbimento dei grassi per il basso pH duodenale. La presenza di ZES dovrebbe essere sempre sospettata in caso di inusuale localizzazione di ulcera, ulcera non responsiva alle terapie standard, diarrea e dimagramento inspiegati. La maggior parte dei gastrinomi associati a MEN 1 dimostrano un’immunoreattività positiva per peptidi diversi dalla gastrina e, pertanto, non determinano l’insorgenza di ZES. 15% 80% Il gastrinoma origina dalle cellule G, localizzate prevalentemente nel duodeno prossimale (83%) e in minor misura nell’antro gastrico, e da popolazioni cellulari denominate D1 a sede pancreatica
 e in minor misura nell’antro gastrico, e da popolazioni cellulari denominate D1 a sede pancreatica; l’espressione genica della gastrina nel pancreas dei mammiferi è limitata alla vita fetale, per cui i gastrinomi pancreatici sono considerati tumori ectopici , mentre i gastrinomi del duodeno, del digiuno e dello stomaco, dove le cellule G sono normalmente presenti, sono considerati tumori eutopici . I gastrinomi rappresentano il tumore neuroendocrino digestivo più frequente nell’ambito della MEN 1: circa il 40% dei casi MEN 1 presenta il gastrinoma e circa il 25% dei casi totali di gastrinoma è associato a MEN 1. Il tumore produce gastrina, un ormone peptidico in grado di stimolare la secrezione acida dello stomaco. Tumori con espressione immunoistochimica di gastrina, senza evidenza clinica di ZES non dovrebbero essere considerati gastrinomi. La sede di più frequente insorgenza è il pancreas, e l’incidenza calcolata è di circa 1/ persone/anno. Sono possibili comunque altre localizzazioni, digestive o addirittura extradigestive. Triangolo del gastrinoma: testa pancreatica-porzione superiore e discendente del duodeno-linfonodi. Il duodeno rappresenta la localizzazione principale dei gastrinomi associati a MEN 1, che sono spesso di piccole dimensioni (<1 cm) e multifocali. Altri siti primari di insorgenza del gastrinoma sono lo stomaco, il digiuno, le vie biliari, fegato, reni, mesenterio, cuore e ovaio; l’ipotesi di insorgenza primaria linfonodale dei gastrinomi è controversa: i gastrinomi duodenali in particolare, infatti, possono metastatizzare quando ancora molto piccoli, suggerendo che i cosiddetti gastrinomi linfonodali siano in realtà metastasi di microgastrinomi duodenali occulti. La malignità riguarda circa il 60% dei casi. Il dolore addominale (78%) e la diarrea (40%) sono le manifestazioni cliniche più importanti. Il dolore addominale è da riferire alla malattia ulcerosa, all’iperperistaltismo e all’infiltrazione neoplastica di rami nervosi. La lesione peptica si localizza nel 75% dei casi nella porzione prossimale del duodeno e meno comunemente nello stomaco distale. La diarrea e la steatorrea sono causate principalmente alla ridotta capacità di assorbimento intestinale per aumentata secrezione gastrica ed all’inattivazione delle lipasi pancreatiche e alla precipitazione de gli acidi biliari con conseguente malassorbimento dei grassi per il basso pH duodenale. La presenza di ZES dovrebbe essere sempre sospettata in caso di inusuale localizzazione di ulcera, ulcera non responsiva alle terapie standard, diarrea e dimagramento inspiegati. La maggior parte dei gastrinomi associati a MEN 1 dimostrano un’immunoreattività positiva per peptidi diversi dalla gastrina e, pertanto, non determinano l’insorgenza di ZES. 15% 80% Il gastrinoma origina dalle cellule G, localizzate prevalentemente nel duodeno prossimale (83%) e in minor misura nell’antro gastrico, e da popolazioni cellulari denominate D1 a sede pancreatica.")
23
Gastrinoma: Clinica spesso multifocali
dimensioni < 1 cm associati a MEN 1 duodenali singolo dimensioni > 1 cm sporadico pancreatici La maggior parte (circa 60%) dei gastrinomi è maligna, più della metà ha già metastatizzato alla diagnosi ed un terzo di casi di ZES, sia sporadici che associati a MEN 1, muore a causa della neoplasia. Il gastrinoma sporadico si localizza a livello pancreatico, mentre il gastrinoma associato a MEN 1 si localizza a livello duodenale ed è spesso di piccole dimensioni e multifocale; la prognosi infausta si associa a lesioni primitive pancreatiche e non duodenali (gastrinoma in MEN 1 va meglio dello sporadico: la mortalità associata a tumore è circa del 38% nei gastrinomi associati a MEN 1 e del 62% in quelli sporadici), sindrome di Cushing ectopica (6% dei casi), alti livelli di gastrina plasmatici, presenza di metastasi ossee (12% dei casi): la causa più frequente di morte è la cachessia neoplastica. Un Ki 67 > 10% è invariabilmente associato allo sviluppo di metastasi. Sebbene la maggior parte dei gastrinomi presenti un andamento maligno, né le dimensioni né le caratteristiche istologiche riflettono con certezza il comportamento biologico della malattia; pertanto i gastrinomi devono essere classificati come tumori a comportamento incerto o come carcinomi endocrini ben differenziati in caso di presenza di angioinvasione e/o metastasi. I gastrinomi con metastasi epatiche sembrano avere un atteggiamento più aggressivo di quelli con metastasi linfonodali soltanto; il rischio di metastatizzazione epatica è correlato alle dimensioni e alla localizzazione pancreatica del tumore primitivo (30% nei pazienti con gastrinoma pancreatico e 3% in quelli con localizzazione duodenale). La progressione di malattia è comunque generalmente lenta, con una sopravvivenza a 5 anni del 65% (62-75%); anche nella malattia metastatica la sopravvivenza è relativamente lunga, con una mediana del 46% a 5 anni per la malattia metastatica linfonodale e del 40% per quella epatica. Pazienti con resezione completa del tumore hanno una sopravvivenza del % a 10 anni. Non esistono markers predittivi di comportamento biologico del gastrinoma. Tuttavia, l’amplificazione di Her2/neu e l’overespressione di EGF ed HGF (Epidermal ed Hepatocyte Growth Factor) sembrano essere associati ad una crescita tumorale di tipo aggressivo. La frequenza di perdita 11q13 e di mutazione del gene MEN 1 è la più elevata di tutti i tumori endocrini pancreatici.
 dei gastrinomi è maligna, più della metà ha già metastatizzato alla diagnosi ed un terzo di casi di ZES, sia sporadici che associati a MEN 1, muore a causa della neoplasia. Il gastrinoma sporadico si localizza a livello pancreatico, mentre il gastrinoma associato a MEN 1 si localizza a livello duodenale ed è spesso di piccole dimensioni e multifocale; la prognosi infausta si associa a lesioni primitive pancreatiche e non duodenali (gastrinoma in MEN 1 va meglio dello sporadico: la mortalità associata a tumore è circa del 38% nei gastrinomi associati a MEN 1 e del 62% in quelli sporadici), sindrome di Cushing ectopica (6% dei casi), alti livelli di gastrina plasmatici, presenza di metastasi ossee (12% dei casi): la causa più frequente di morte è la cachessia neoplastica. Un Ki 67 > 10% è invariabilmente associato allo sviluppo di metastasi. Sebbene la maggior parte dei gastrinomi presenti un andamento maligno, né le dimensioni né le caratteristiche istologiche riflettono con certezza il comportamento biologico della malattia; pertanto i gastrinomi devono essere classificati come tumori a comportamento incerto o come carcinomi endocrini ben differenziati in caso di presenza di angioinvasione e/o metastasi. I gastrinomi con metastasi epatiche sembrano avere un atteggiamento più aggressivo di quelli con metastasi linfonodali soltanto; il rischio di metastatizzazione epatica è correlato alle dimensioni e alla localizzazione pancreatica del tumore primitivo (30% nei pazienti con gastrinoma pancreatico e 3% in quelli con localizzazione duodenale). La progressione di malattia è comunque generalmente lenta, con una sopravvivenza a 5 anni del 65% (62-75%); anche nella malattia metastatica la sopravvivenza è relativamente lunga, con una mediana del 46% a 5 anni per la malattia metastatica linfonodale e del 40% per quella epatica. Pazienti con resezione completa del tumore hanno una sopravvivenza del % a 10 anni. Non esistono markers predittivi di comportamento biologico del gastrinoma. Tuttavia, l’amplificazione di Her2/neu e l’overespressione di EGF ed HGF (Epidermal ed Hepatocyte Growth Factor) sembrano essere associati ad una crescita tumorale di tipo aggressivo. La frequenza di perdita 11q13 e di mutazione del gene MEN 1 è la più elevata di tutti i tumori endocrini pancreatici.")
24
Test genetico La MEN1 dovrebbe essere sospettata nei pazienti con: iperparatiroidismo ad esordio <30 anni o a base multighiandolare o con elevata incidenza familiare; tumori endocrini del pancreas multifocali; Zollinger-Ellison; due endocrinopatie di pancreas, ipofisi, paratiroidi Test genetico disponibile; utile in fase precoce per il monitoraggio delle successive endocrinopatie

25
MEN1 Screening: when and how

26
Therapeutic considerations
Despite its earlier recognition, MEN 1 is the most challenging of the MEN syndromes. Each affected patient can be expected to undergo at least two or more surgical procedures; it is necessary to recognize the high probability of recurrent or new neoplasms in potentially affected organ systems and to balance this likelihood against the possible side effects of intervention, such as Hypoparathyroidism Hypopituitarism Endocrine and exocrine pancreatic insufficency Sindrome neoplastica quasi sempre assente in tutti i neuroendocrini ad eccezione dei glucagonomi. Invasione locale presentazione tipica dei NE non secernenti LAVORO DEL CHIRURGO! Sindrome endocrina prognosi peggiore ad eccezione degli insulinomi LAVORO DEL CLINICO!

27
MULTIPLE ENDOCRINE NEOPLASIA
MEN TYPE 2

28
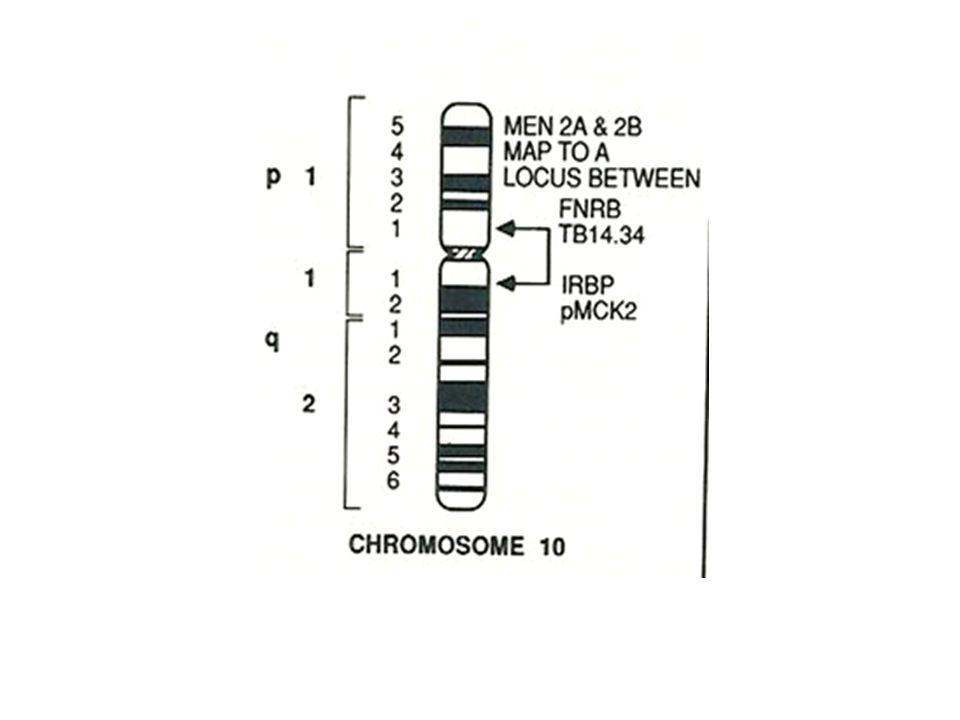
Overview The hallmark of MEN2 is a very high lifetime risk of developing medullary thyroid carcinoma (MTC) more than 95% in untreated patients. Three clinical subtypes MEN2A, MEN2B, and familial MTC (FMTC) have been defined based on the risk of: pheochromocytoma hyperparathyroidism the presence or absence of characteristic physical features The prevalence of MEN2 has been estimated at 1 in 35,000 individuals
 have been defined based on the risk of: pheochromocytoma. hyperparathyroidism. the presence or absence of characteristic physical features. The prevalence of MEN2 has been estimated at 1 in 35,000 individuals.")
29
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2
Definition The MEN 2 syndrome has been sub-categorized into two variants called MEN 2A and MEN 2B (formerly MEN 3)
")
30
MEN 2 and its clinical variants or syndromes

31
MEN 2 and its clinical variants or syndromes

32
MEN 2: genetica e fisiopatologia
Mutation of the c-ret protooncogene have been identified in 93 to 95% of pts with MEN 2. Two regions of the Ret tyrosine kinase receptor are mutated

34
MEN TYPE 2: diagram of the c-ret protoncogene

35
Percent of MEN 2 Clinical Features by Subtype
Medullary Thyroid Carcinoma Pheochromo cytoma Parathyroid Disease MEN 2A 95% 50% 20-30% FMTC 100% 0% 0% MEN 2B 100% 50% Uncommon

36
Testing Used in MEN 2 Mutation Detection Rate Clinical Testing Test
Type Test Availability MEN 2A 95% DNA-based Clinical Testing FMTC 85% DNA-based MEN 2B 95% DNA-based

37
Screening and Risk assessment
MEN2 accounts for approximately 25% of all cases of MTC and approximately 7% of individuals presenting with apparently sporadic MTC. RET genetic testing is considered the standard of care for newly identified MTC patients, regardless of age at diagnosis or family history. The identification of a mutation provides essential risk information for the patient’s family members, and genotype-phenotype correlations can help estimate the patient’s risk of developing additional endocrinopathies (eg, pheochromocytoma, primary hyperparathyroidism), provide prognostic information, and guide the surgical management of MTC.
, provide prognostic information, and guide the surgical management of MTC.")
38
MEN 2A: Overview The MEN 2A syndrome consist of
multifocal medullary thyroid carcinoma unilateral or bilateral pheochromocytoma parathyroid hyperplasia or adenoma Approximately 4% to 5% of cases of apparently sporadic pheochromocytoma occurring before age 50 years are due to mutations of RET and are thus associated with MEN2A. MEN2A-associated pheochromocytomas almost always secrete epinephrine and may or may not secrete norepinephrines. In addition, malignancy and extra-adrenal location are extremely rare in MEN2A.

39
MEN 2A: Clinical features
Patients with this syndrome can present with manifestations of a pheochromocytoma, a thyroid nodule, hypercalcemia or some combination of the three, but at present the routine screening of affected families makes early thyroid C-cell hyperplasia (elevation of circulating calcitonin), the most common initial presentation (followed by Pheochromocytoma in about half pts and parathyroid abnormalities in 10 to 35%).
, the most common initial presentation (followed by Pheochromocytoma in about half pts and parathyroid abnormalities in 10 to 35%).")
40
MEN 2A Screening: when and how

41
MEN 2A: Clinical features
Medullary thyroid carcinoma (MTC) Multicentric neoplasm of parafollicular or C cell of thyroid gland. The first demonstrable abnormality is hyperplasia of C cells followed by Histological progression: nodular hyperplasia microscopic medullary thyroid carcinoma frank medullary thyroid carcinoma The time required for this progression through these histologic stages, is not known but the process may require decades.
 Multicentric neoplasm of parafollicular or C cell of thyroid gland. The first demonstrable abnormality is hyperplasia of C cells followed by. Histological progression: nodular hyperplasia. microscopic medullary thyroid carcinoma. frank medullary thyroid carcinoma. The time required for this progression through these histologic stages, is not known but the process may require decades.")
42
MEN 2A: Clinical features
Pheochromocytoma Adrenal chromaffin tissue undergoes the same type of histological progression as that observed for C cell Histological progression: - hyperplasia of chromaffin cells - nodular hyperplasia - pheochromocytoma

43
MEN 2A: Clinical features
Pheochromocytoma Diagnosis confirmed by: Biochemical features CT MRI Scanning with MIBG

44
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2 B
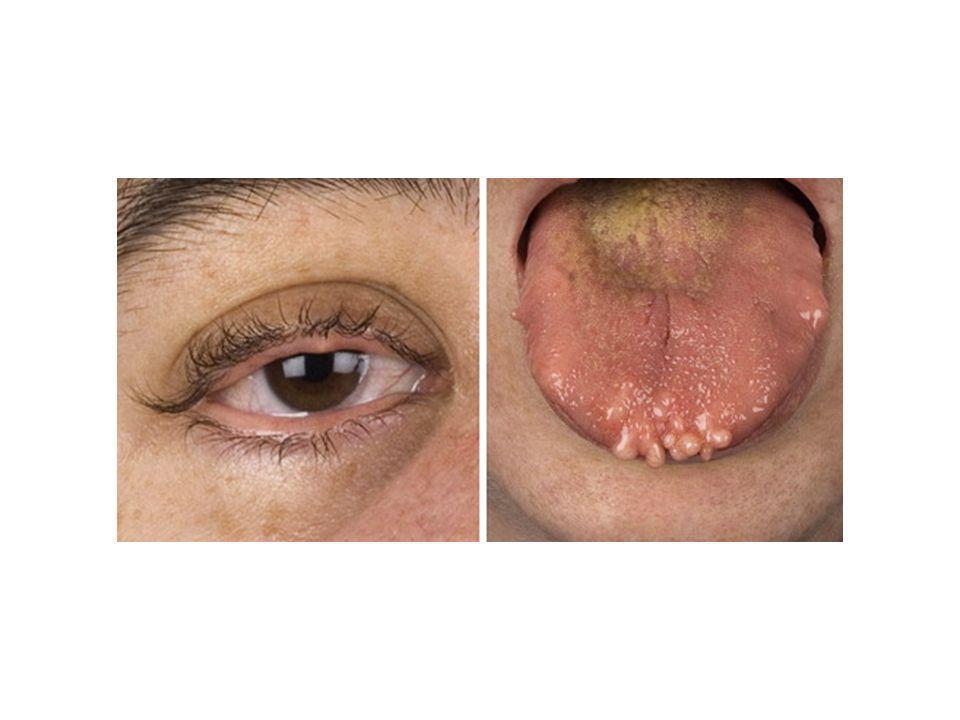
Introduction The MEN 2B (or MEN 3) syndrome consist of multifocal medullary thyroid carcinoma unilateral or bilateral pheochromocytoma multiple mucosal neuromas marfanoid habitus The hallmark of this syndrome is the presence of characteristic mucosal neuromas on the distal portion of the tongue, the lips and subconjunctival areas and throught the gastrintestinal tract.
 syndrome consist of. multifocal medullary thyroid carcinoma. unilateral or bilateral pheochromocytoma. multiple mucosal neuromas. marfanoid habitus. The hallmark of this syndrome is the presence of characteristic mucosal neuromas on the distal portion of the tongue, the lips and subconjunctival areas and throught the gastrintestinal tract.")
45
Multiple Mucosal Neuromas

46
Multiple Mucosal Neuromas

47
Multiple Mucosal Neuromas

49
MULTIPLE ENDOCRINE NEOPLASIA TYPE 2 B
The clinical course of patients with medullary thyroid carcinoma in MEN 2B is more aggressive than that in MEN 2A. Metastatic disease can occur in children younger than 1 year age and there is shorter average survival time in patients with metastatic disease. The identification of mucosal neuroma phenotype in a child should alert the physician to the diagnosis of medullary thyroid carcinoma

50
MEN 2B Screening: when and how

Presentazioni simili
 Università degli Studi, Pavia (founder) IRCCS Fondazione.>")





>")












