Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
1) Algoritmi di allineamento 2) Algoritmi di ricerca in database
 Algoritmi di allineamento 2) Algoritmi di ricerca in database")
2
similarità allineamento
abbiamo visto che per generare e valutare il miglior allineamento di due sequenze di lunghezza m e n, è necessario effettuare m x n confronti il numero di operazioni da effettuare cresce e i tempi di calcolo di conseguenza si allungano se si vogliono considerare anche i possibili gap in tutte le posizioni (e di tutte le lunghezze possibili) di entrambe le sequenze IPLMTRWDQEQESDFGHKLPIYTREWCTRG |||||||||| CHKIPLMTRWDQQESDFGHKLPVIYTREW IPLMTRWDQEQESDFGHKLP-IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVIYTREW
 di entrambe le sequenze. IPLMTRWDQEQESDFGHKLPIYTREWCTRG |||||||||| CHKIPLMTRWDQQESDFGHKLPVIYTREW. IPLMTRWDQEQESDFGHKLP-IYTREWCTRG ||||||||| |||||||||| |||||| CHKIPLMTRWDQ-QESDFGHKLPVIYTREW.")
3
noi VOGLIAMO considerare i gap, ma non POSSIAMO permetterci algoritmi che considerino tutti i possibili gap in tutte le possibili posizioni gli allineamenti possono essere visualizzati graficamente in modo rapido (con algoritmi dell’ordine di mxn) con matrici di punti (dot plots) identità finestra = 5 finestra = 15
 con matrici di punti (dot plots) identità. finestra = 5. finestra = 15.")
4
abbiamo definito un modo sofisticato di valutare il punteggio di un allineamento, che tenga conto di
punteggi diversi per ogni possibile coppia di residui allineati (matrici di sostituzione) gap penalties e gap extension penalties per valutare penalizzazioni dovute all’introduzione di gap nelle sequenze allineate
 gap penalties e. gap extension penalties per valutare penalizzazioni dovute all’introduzione di gap nelle sequenze allineate.")
5
PROGRAMMAZIONE DINAMICA
che algoritmi possiamo usare? razionalizzare il problema: considerare anche i gap pur continuando ad utilizzare un algoritmo dell’ordine di n2) PROGRAMMAZIONE DINAMICA
 PROGRAMMAZIONE DINAMICA.")
6
. spesso l’output mostra più allineamenti
spesso l’output mostra più allineamenti DIVERSI col massimo del punteggio ci mette TROPPO TEMPO per effettuare una ricerca esaustiva

7
. scegliere una matrice di sostituzione per
scegliere una matrice di sostituzione per valutare gli appaiamenti tra residui definire dei punteggi di penalizzazione per i gaps algoritmi di allineamento che utilizzano una tecnica di programmazione dinamica: Needleman e Wunsch (1970) Smith e Waterman (1981)
 Smith e Waterman (1981)")
8
3 passi successivi 1) consideriamo le due sequenze da allineare in una specie di dot plot : nelle caselle scriviamo i punteggi in rosso derivati dalla matrice di sostituzione scelta
 consideriamo le due sequenze da allineare in una specie di dot plot : nelle caselle scriviamo i punteggi in rosso derivati dalla matrice di sostituzione scelta.")
10
QERTY |||: QERS Q E R T Y Q 4 2 1 -1 -4 E 2 4 -1 -4 R 1 -1 6 -1 -4 S
allineamento punteggio QERTY |||: QERS QQ + EE + RR + TS = = 15 cominciamo a calcolare la matrice di questo allineamento usata nell’algoritmo di programmazione dinamica Q E R T Y Q 4 2 1 -1 -4 E 2 4 -1 -4 R 1 -1 6 -1 -4 S -1 1 -3

11
2) ricerca del percorso che consente di ottenere il massimo punteggio in base a delle regole stabilite, tenendo anche conto dei gaps se una sequenza è scritta da sinistra a destra e l’altra dall'alto in basso, allora qualsiasi percorso valido deve mantenere sempre una direzione tendenziale che va dall'angolo in alto a sinistra a quello in basso a destra

12
Misura della similarità:
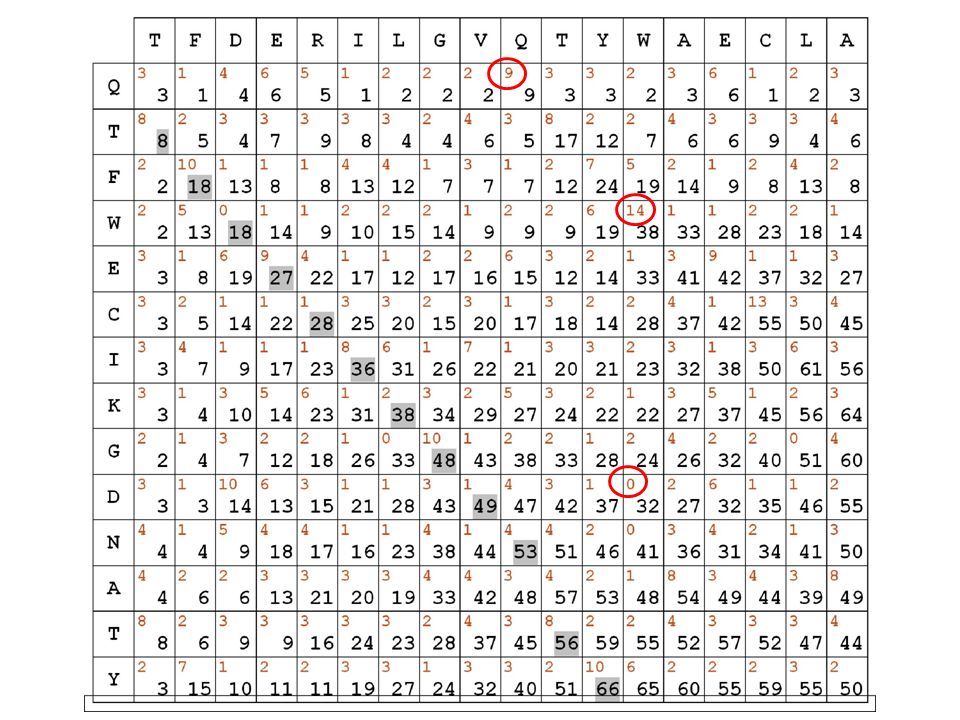
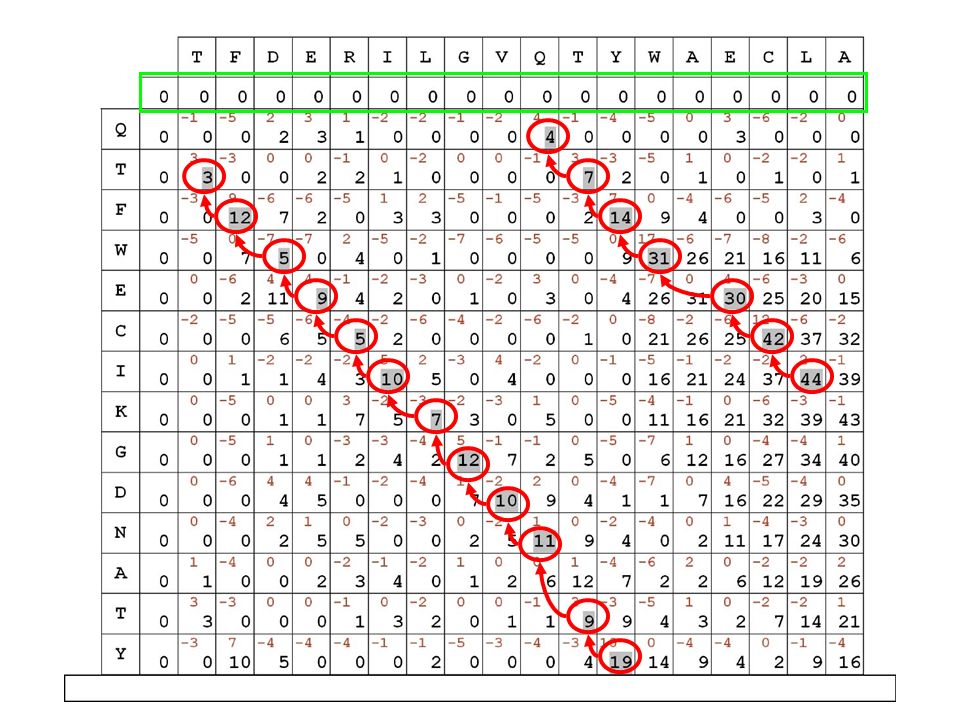
i punteggi in diagonale si SOMMANO fuori dalla diagonale, si PENALIZZA di 5 punti evidenzieremo in grigio il migliore percorso all’interno della matrice, secondo le regole e i punteggi stabiliti

14
il migliore allineamento globale per le sequenze in matrice risulta quindi il seguente:
TFDERILGVQ-TYWAECLA || | | | . || QTFWECIKGDNATY

15
TFDERILGVQ-TYWAECLA || | | | . || QTFWECIKGDNATY
per ricostruire l’allineamento migliore si può memorizzare il percorso disegnato riempiendo la matrice TFDERILGVQ-TYWAECLA || | | | . || QTFWECIKGDNATY alla fine bisogna partire dalla casella a punteggio maggiore e ricostruire il percorso a ritroso seguendo le freccette

16
Qual è il tipo di allineamento che consideriamo più interessante dal punto di vista biologico?
date due sequenze, per esempio: 1) LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK 2) TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG
 LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK 2) TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG.")
17
Allineamento globale o locale?
LTGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||. | | | .| .| || || | || TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG Allineamento locale: TGARDWEDIPLWTDWDIEQESDFKTRAFGTANCHK ||||||||.||||| TGIPLWTDWDLEQESDNSCNTDHYTREWGTMNAHKAG

18
Allineamento globale o locale?
a livello di DNA, troviamo regioni con similarità locali che riflettono situazioni interessanti: ad esempio introni/esoni, inserzioni/delezioni, transposoni, regioni promotore… esoni introni

19
allineamenti globali:
confrontare accuratamente due sequenze la cui similarità sia estesa per tutta la lunghezza (es. Proteine omologhe)
")
20
Usando matrici di sostituzione contenenti esclusivamente valori positivi il valore massimo della matrice si trova sempre nell’ultima riga o nell’ultima colonna ne consegue che l’allineamento ottenuto è un allineamento globale il numero di operazioni effettuate è proporzionale a m x n abbiamo considerato tutti i possibili allineamenti tra le due sequenze e tutte le possibili posizioni per i gap

21
Come ottenere un allineamento locale?
se le matrici contenessero sia valori positivi che negativi (come le PAM o le BLOSUM), i valori più alti potrebbero trovarsi anche in porzioni INTERNE alla matrice ogni percorso che totalizza un punteggio negativo viene azzerato e può venire considerato come l’inizio di un nuovo allineamento locale quando i tre percorsi di possibile provenienza per una casella risultano negativi, il punteggio associato alla casella sarà pari a zero
, i valori più alti potrebbero trovarsi anche in porzioni INTERNE alla matrice. ogni percorso che totalizza un punteggio negativo viene azzerato e può venire considerato come l’inizio di un nuovo allineamento locale. quando i tre percorsi di possibile provenienza per una casella risultano negativi, il punteggio associato alla casella sarà pari a zero.")
22
3 -3 9 12
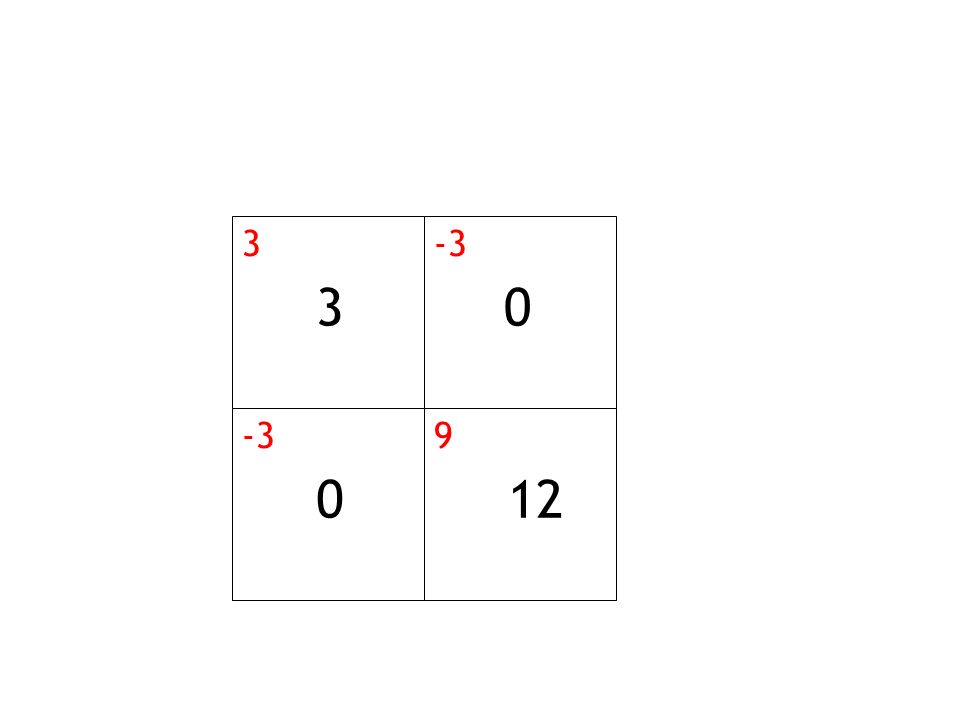
23
-7 -6 -2 1
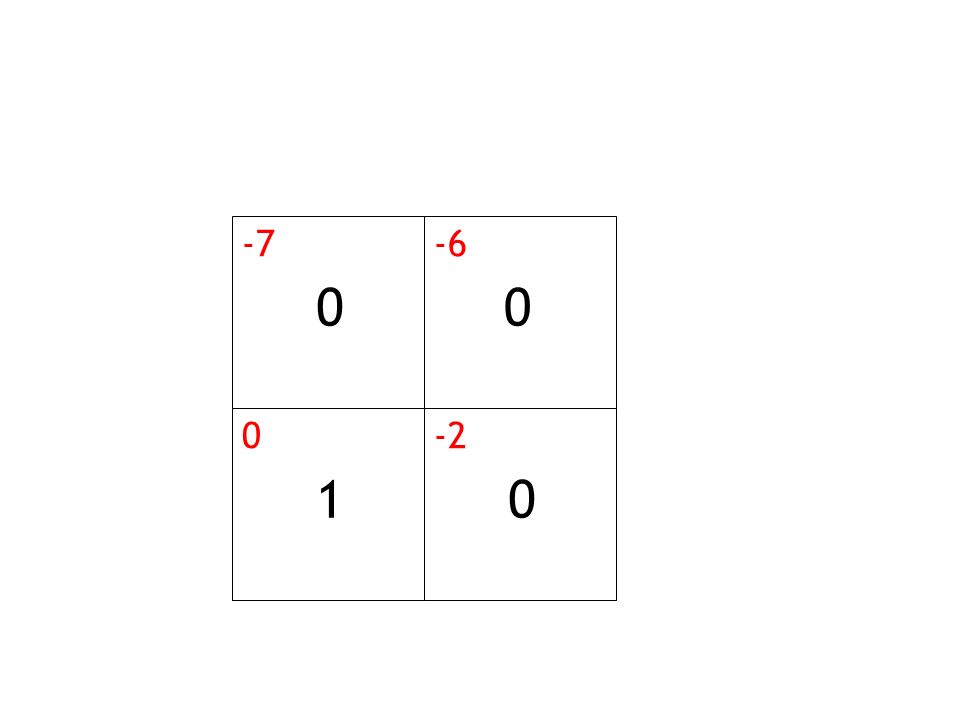
25
Gli algoritmi per il Database similarity searching
FASTA BLAST

26
Prerequisiti dei metodi di Database similarity searching
Velocità Sensibilità Selettività

27
Prerequisiti dei metodi di Database similarity searching
Fissata una soglia minima di similarità se TP = positivi veri PP = positivi predetti (falsi e veri) AP = actual positives ( n.reale di sequenze omologhe presenti nel db indipendentemente dalla soglia)
 AP = actual positives ( n.reale di sequenze omologhe presenti nel db indipendentemente dalla soglia)")
28
Database similarity searching
Sensibilità = TP/AP Selettività =TP/PP La scelta della soglia e’ fatta su basi statistiche ed e’ determinante. Alta sensibilità bassa selettività e viceversa

29
Database similarity searching
Ambedue i metodi (FASTA e BLAST) effettuano ricerche di similarità mediante applicazioni di metodi per allineamenti locali e, dal confronto di una sequenza anonima con set di sequenze a funzione nota, selezionano le sequenze con punteggi di similarità (scores) superiore ad una certa soglia (threshold), valutata su basi statistiche e dinamicamente in correlazione con il dataset sotto studio.
 effettuano ricerche di similarità mediante applicazioni di metodi per allineamenti locali e, dal confronto di una sequenza anonima con set di sequenze a funzione nota, selezionano le sequenze con punteggi di similarità (scores) superiore ad una certa soglia (threshold), valutata su basi statistiche e dinamicamente in correlazione con il dataset sotto studio.")
30
Database searching Ambedue i metodi ottengono una elevata velocità di esecuzione grazie alla trasformazione delle sequenze in vettori Scelta la dimensione w della stringa su cui operare per la ricerca delle similarità, le sequenze da confrontare sono trasformate in vettori di dimensione Dw, in ogni cella dei quali sono riportate le posizioni in cui la i-esima "stringa" di dimensione w ricorre nella sequenza

31
Database searching FASTA
Ammette i gaps e produce una sola regione di miglior similarità per ogni coppia analizzata (query seq. vs. db seq)
")
32
Database searching BLAST
Non ammette i gaps e produce più regioni di similarità per ogni coppia analizzata

33
FASTA Fissato il parametro ktup ( il numero di residui contigui identici), consideriamo due sequenze, S1 e S2, e cerchiamo le k-tuple perfettamente identiche il cui inizio é localizzato rispettivamente nelle posizioni i e j; nell'ipotetica matrice dot-plot relativa a queste due sequenze tale identità sarà localizzata nella diagonale m=i-j.
, consideriamo due sequenze, S1 e S2, e cerchiamo le k-tuple perfettamente identiche il cui inizio é localizzato rispettivamente nelle posizioni i e j; nell ipotetica matrice dot-plot relativa a queste due sequenze tale identità sarà localizzata nella diagonale m=i-j.")
34
FASTA Vengono individuate le diagonali che presentano la più alta densità di k-tuple identiche. Le k-tuple identiche vengono assegnate alla medesima regione di similarità se sono separate da un numero di residui inferiore ad un valore soglia prestabilito nell’algoritmo (Pearson, 1990). Vengono selezionate le prime dieci regioni di similarità localizzate anche su differenti diagonali.
. Vengono selezionate le prime dieci regioni di similarità localizzate anche su differenti diagonali.")
35
FASTA fase 2 Il punteggio di similarità relativo alle regioni precedentemente considerate viene calcolato prendendo in considerazione tratti di sequenza identici più corti di ktup, eventuali sostituzioni conservative e consentendo anche l’introduzione di piccoli gaps. Per le sequenze proteiche PAM-250 Per le sequenze nucleotidiche punteggio positivo alle identità e negativo alle differenze. Tra tutte le regioni diagonali di similarità quella che totalizza il massimo punteggio di similarità viene definita "regione primaria di similarità" e il punteggio corrispondente é denominato init1

36
FASTA fase 3 Le regioni di similarità precedentemente determinate vengono potenzialmente ricongiunte per formare un unico allineamento. Questa ricongiunzione viene effettuata se la penalità di ricongiungimento, proporzionale alla distanza tra le regioni di similarità, é inferiore al contributo dato al punteggio di similarità dalla regione di similarità che viene ricongiunta nell'allineamento.

37
FASTA fase 3 Il punteggio di similarità relativo all'allineamento che così viene determinato viene denominato punteggio initn e viene usato per ordinare le sequenze della banca dati secondo il grado di similarità decrescente con la sequenza sonda in esame.

38
FASTA fase 4 Nella quarta ed ultima fase, l'allineamento precedentemente ottenuto viene ulteriormente ottimizzato utilizzando la procedura di allineamento descritta da Chao et al. (1992) che utilizza un algoritmo per l'allineamento di due sequenze all'interno di una banda diagonale di dimesioni predeterminate. Il punteggio di similarità calcolato in quest'ultima fase viene denominato punteggio opt.
 che utilizza un algoritmo per l allineamento di due sequenze all interno di una banda diagonale di dimesioni predeterminate. Il punteggio di similarità calcolato in quest ultima fase viene denominato punteggio opt.")
39
BLAST (Basic Local Alignment Search Tool)
Le coppie di segmenti, presenti nella stessa coppia di sequenze, che totalizzino un punteggio di similarità statisticamente significativo, superiore ad una soglia S, vengono definiti HSP (High scoring Segment Pairs). Nella stessa coppia possono esserci più HSP di cui é anche possibile calcolare la probabilità di occorrenza (Karlin & Altschul, 1993).
. Nella stessa coppia possono esserci più HSP di cui é anche possibile calcolare la probabilità di occorrenza (Karlin & Altschul, 1993).")
40
BLAST (Basic Local Alignment Search Tool)
Una volta individuata una stringa coincidente (matching word), questa viene estesa verso entrambe le direzioni. La procedura di estensione viene arrestata in una delle due direzioni quando il punteggio di similarità relativo alla coppia di segmenti in esame viene decrementato, di un certo valore prestabilito, rispetto a quello relativo ad una minore estensione della stessa coppia di segmenti.
, questa viene estesa verso entrambe le direzioni. La procedura di estensione viene arrestata in una delle due direzioni quando il punteggio di similarità relativo alla coppia di segmenti in esame viene decrementato, di un certo valore prestabilito, rispetto a quello relativo ad una minore estensione della stessa coppia di segmenti.")
42
BLAST (Basic Local Alignment Search Tool)
L’idea di fondo : l’allineamento ottimale tra due sequenze omologhe, anche se altamente divergenti, ha un’alta probabilità di contenere una o più coppie di segmenti caratterizzati da un punteggio di similarità piuttosto elevato e tale da consentire una affidabile distinzione tra sequenze correlate da relazione di omologia e sequenze non correlate. Un algoritmo in grado di individuare in modo estremamente rapido tutte le coppie di segmenti per cui il punteggio di similarità risulta statisticamente significativo.

43
BLAST (Basic Local Alignment Search Tool)
La peculiare strategia di BLAST é di cercare esclusivamente coppie di segmenti che contengano una coppia di “stringhe” di lunghezza w il cui relativo punteggio di similarità sia almeno pari ad una soglia T. Il valore della soglia T (≤S) viene stimato sulla base della dimensione di w.
 viene stimato sulla base della dimensione di w.")
44
BLAST (Basic Local Alignment Search Tool)
S(a,b) utilizza matrici BLOSUM o PAM per le sequenze aminoacidiche o un punteggio pari a +5 per le identità e -4 per le differenze per sequenze nucleotidiche Si definisce MSP (Maximal scoring Segment Pair) la coppia di segmenti, di eguale lunghezza, che realizza il massimo punteggio di similarità nel confronto di due sequenze; l’algorimo ne valuta in modo rigoroso la significatività statistica (Karlin & Altschul, 1990, 1993).
 utilizza matrici BLOSUM o PAM per le sequenze aminoacidiche o un punteggio pari a +5 per le identità e -4 per le differenze per sequenze nucleotidiche. Si definisce MSP (Maximal scoring Segment Pair) la coppia di segmenti, di eguale lunghezza, che realizza il massimo punteggio di similarità nel confronto di due sequenze; l’algorimo ne valuta in modo rigoroso la significatività statistica (Karlin & Altschul, 1990, 1993).")
Presentazioni simili















>")






