Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Basi cellulari della farmacocinetica

2
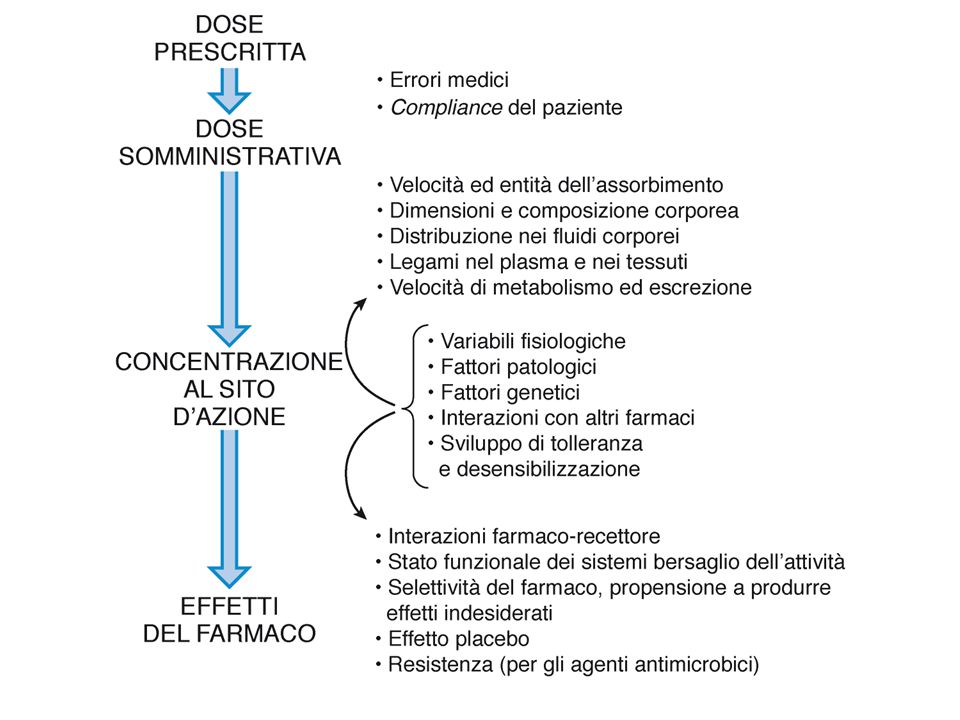
FARMACOCINETICA Branca della farmacologia che studia l’andamento temporale delle concentrazioni di farmaco all’interno di un organismo, al fine di prevedere con ragionevole accuratezza la concentrazione del farmaco sul sito d’azione che sarà prodotta nel paziente da un determinato regime terapeutico.

4
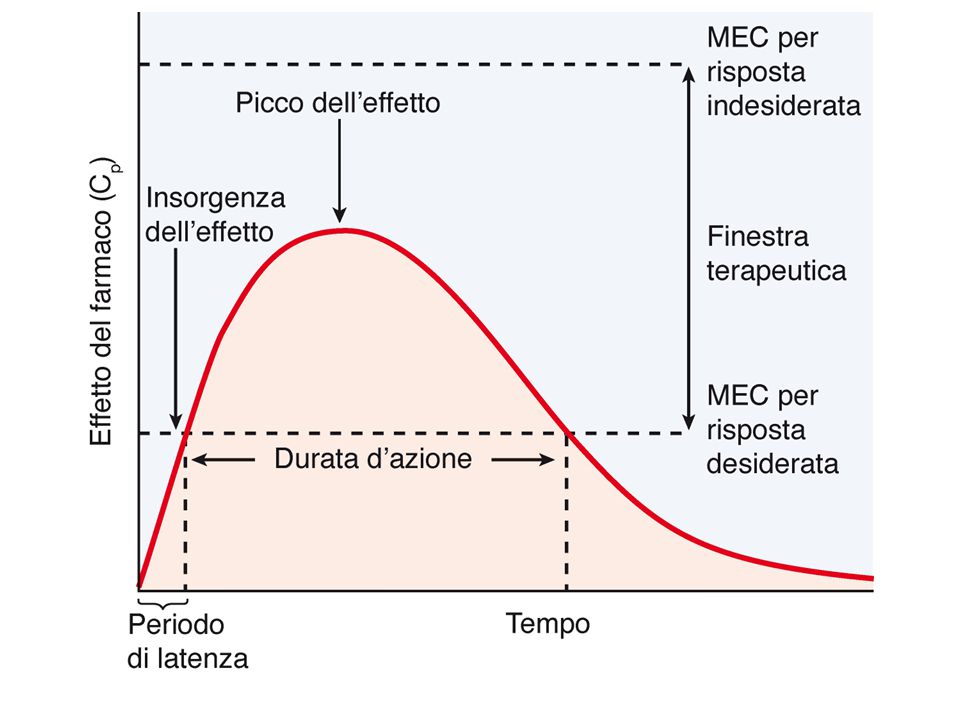
L’entità delle risposte ad un farmaco è funzione della sua concentrazione sul sito d’azione: dosi troppo basse produrranno risposte inadeguate, mentre dosi troppo elevate potrebbero indurre effetti indesiderati tali da vanificare l’efficacia della terapia. Tra questi due limiti, che definiscono la finestra terapeutica, l’intervento farmacologico è corretto. Obiettivo del medico: rientrare nella FT scegliendo opportune dosi, vie e frequenza di somministr (compito difficile per F con IT basso)
")
5
L’indice terapeutico è un indice della maneggevolezza di un farmaco

7
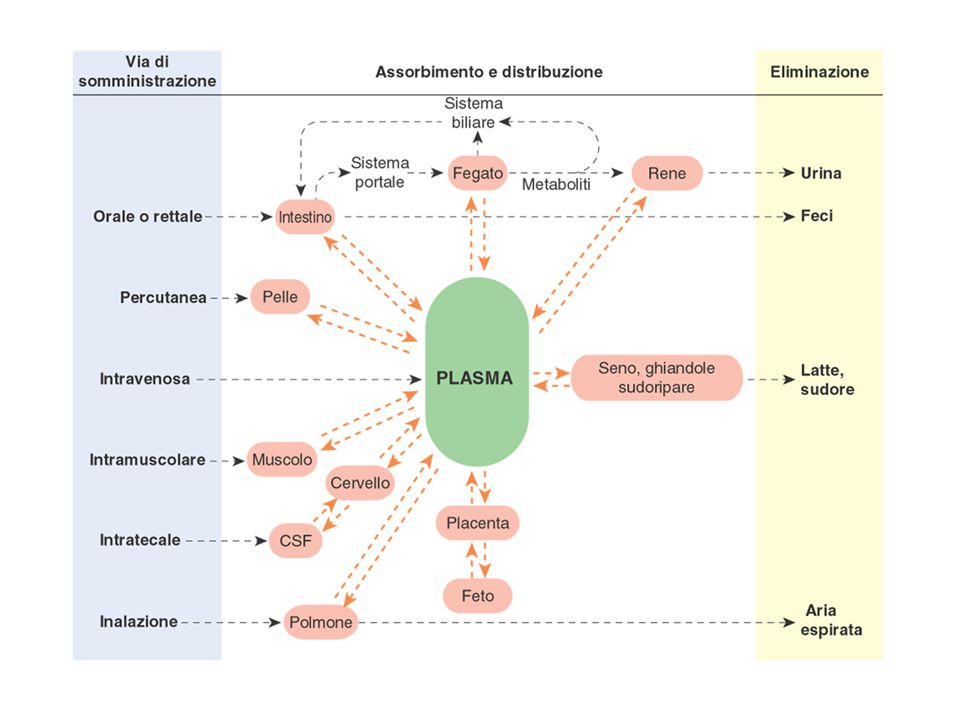
Il viaggio del farmaco nell’organismo:
assorbimento, distribuzione, eliminazione

8
ASSORBIMENTO: la serie di processi che consentono il passaggio del farmaco dal sito di somministrazione al circolo ematico. Vie di somministrazione interstiziali (i.m. o s.c.): il F deve diffondere dal sito di iniezione e superare l’ostacolo delle cellule endoteliali. Altre vie di somministrazione (orale, rettale, sublinguale, transcutanea, etc.): il F deve superare anche la barriera epiteliale che separa l’interstizio dall’esterno. In alcuni casi il farmaco si somministra direttamente nel sito d’azione (es. antiacidi, anestetici locali somministrati nei liquidi interstiziali della regione interessata) o per via endovenosa per cui non c’è assorbimento.
: il F deve diffondere dal sito di iniezione e superare l’ostacolo delle cellule endoteliali. Altre vie di somministrazione (orale, rettale, sublinguale, transcutanea, etc.): il F deve superare anche la barriera epiteliale che separa l’interstizio dall’esterno. In alcuni casi il farmaco si somministra direttamente nel sito d’azione (es. antiacidi, anestetici locali somministrati nei liquidi interstiziali della regione interessata) o per via endovenosa per cui non c’è assorbimento.")
9
DISTRIBUZIONE: diffusione del farmaco nell’organismo (sangue -> tutti gli organi: organo bersaglio e altri tessuti). La velocità di distribuzione dipende dal diverso grado di irrorazione dei vari organi/tessuti (cervello > muscolo > tessuto adiposo). Una volta raggiunto l’equilibrio, la concentrazione del farmaco nei vari tessuti può essere molto diversa da quella ematica (“tropismo” del F per uno specifico organo o tessuto). Il passaggio nei liquidi interstiziali avviene a livello capillare e può essere influenzato dalla permeabilità del letto vascolare e dalle dimensioni/caratteristiche del F. La permeabilità dei vasi può dipendere da: organizzazione anatomica della rete capillare [differisce nei vari distretti (es. SNC e placenta)] presenza di processi patologici (es. infiammazione). La membrana basale dell’endotelio rende la diffusione estremamente limitata per: sostanze con PM ≥ albumina (eccetto per i sinusoidi epatici) legame del F alle proteine plasmatiche o agli elementi corpuscolati del sangue.
. Una volta raggiunto l’equilibrio, la concentrazione del farmaco nei vari tessuti può essere molto diversa da quella ematica ( tropismo del F per uno specifico organo o tessuto). Il passaggio nei liquidi interstiziali avviene a livello capillare e può essere influenzato dalla permeabilità del letto vascolare e dalle dimensioni/caratteristiche del F. La permeabilità dei vasi può dipendere da: organizzazione anatomica della rete capillare [differisce nei vari distretti (es. SNC e placenta)] presenza di processi patologici (es. infiammazione). La membrana basale dell’endotelio rende la diffusione estremamente limitata per: sostanze con PM ≥ albumina (eccetto per i sinusoidi epatici) legame del F alle proteine plasmatiche o agli elementi corpuscolati del sangue.")
10
ELIMINAZIONE: biotrasformazione ed escrezione del farmaco dall’organismo.
La biotrasformazione avviene principalmente a livello epatico. L’escrezione può avvenire a livello di: Reni -> Un’alta frazione del flusso plasmatico sistemico è diretta al rene e attraversa il filtro glomerulare. Circa il 4% di tutto il plasma passa nel tubulo renale ogni minuto e con esso il farmaco (se non legato alle proteine) che viene così eliminato (o riassorbito). Polmoni -> l’eliminazione alveolare è importante soprattutto per i gas Sudore Bile -> una parte del farmaco può essere riassorbita a livello intestinale (ricircolo entero-epatico) Con il passare del tempo, la conc del farmaco nell’organismo diminuisce, per cui è importante comprendere le cinetiche di eliminazione nel caso di terapie prolungate (ogni dose si somma al F “residuo” dalla somministrazione precedente). NB: i processi di eliminazione possono essere fortemente influenzati dall’età, da diverse patologie e dall’assunzione concomitante di altri farmaci/sostanze.
 che viene così eliminato (o riassorbito). Polmoni -> l’eliminazione alveolare è importante soprattutto per i gas. Sudore. Bile -> una parte del farmaco può essere riassorbita a livello intestinale (ricircolo entero-epatico) Con il passare del tempo, la conc del farmaco nell’organismo diminuisce, per cui è importante comprendere le cinetiche di eliminazione nel caso di terapie prolungate (ogni dose si somma al F residuo dalla somministrazione precedente). NB: i processi di eliminazione possono essere fortemente influenzati dall’età, da diverse patologie e dall’assunzione concomitante di altri farmaci/sostanze.")
13
PASSAGGIO DEI FARMACI ATTRAVERSO LE MEMBRANE CELLULARI
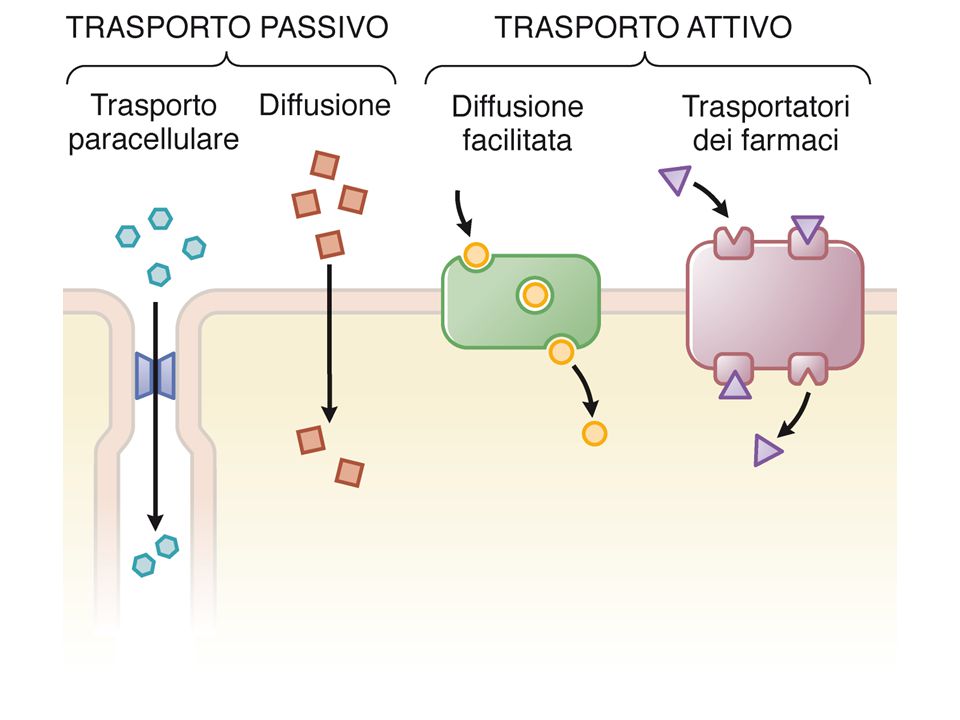
I processi di assorbimento, distribuzione, metabolismo ed eliminazione sono tutti influenzati dalla capacità del farmaco di attraversare le membrane cellulari. Per es., a seguito di somministrazione orale, il F deve attraversare l’epitelio gastro-intestinale, penetrare nel circolo linfatico/ematico e lasciarlo in un altro punto dell’organismo, eventualmente superando altre barriere (es. BEE). Intanto il F è sottoposto all’azione metabolica di enzimi circolanti, ma se è capace di penetrare all’interno delle cellule viene metabolizzato da enzimi intracellulari (es. a livello epatico) o addirittura può essere trasportato dalle cellule (es. azitromicina). Anche l’escrezione renale dipende dalla capacità del farmaco di essere riassorbito a livello dell’epitelio tubulare. Tre modalità di passaggio: Diffusione passiva Endo-esocitosi Trasporto proteina-mediato (carriers, simporti, antiporti e pompe)
. Intanto il F è sottoposto all’azione metabolica di enzimi circolanti, ma se è capace di penetrare all’interno delle cellule viene metabolizzato da enzimi intracellulari (es. a livello epatico) o addirittura può essere trasportato dalle cellule (es. azitromicina). Anche l’escrezione renale dipende dalla capacità del farmaco di essere riassorbito a livello dell’epitelio tubulare. Tre modalità di passaggio: Diffusione passiva. Endo-esocitosi. Trasporto proteina-mediato (carriers, simporti, antiporti e pompe)")
15
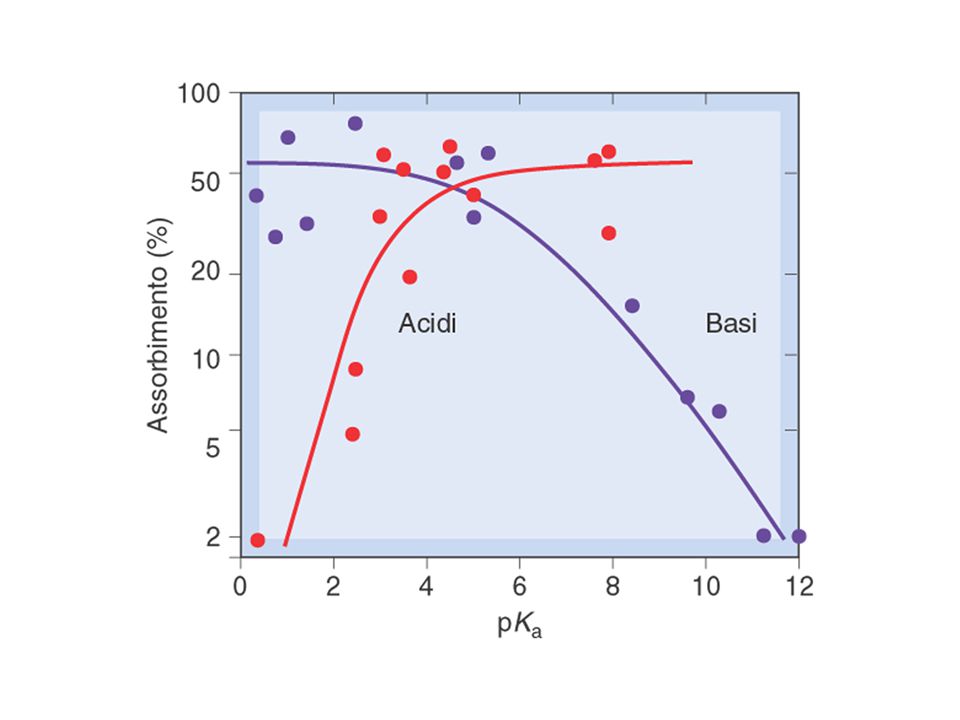
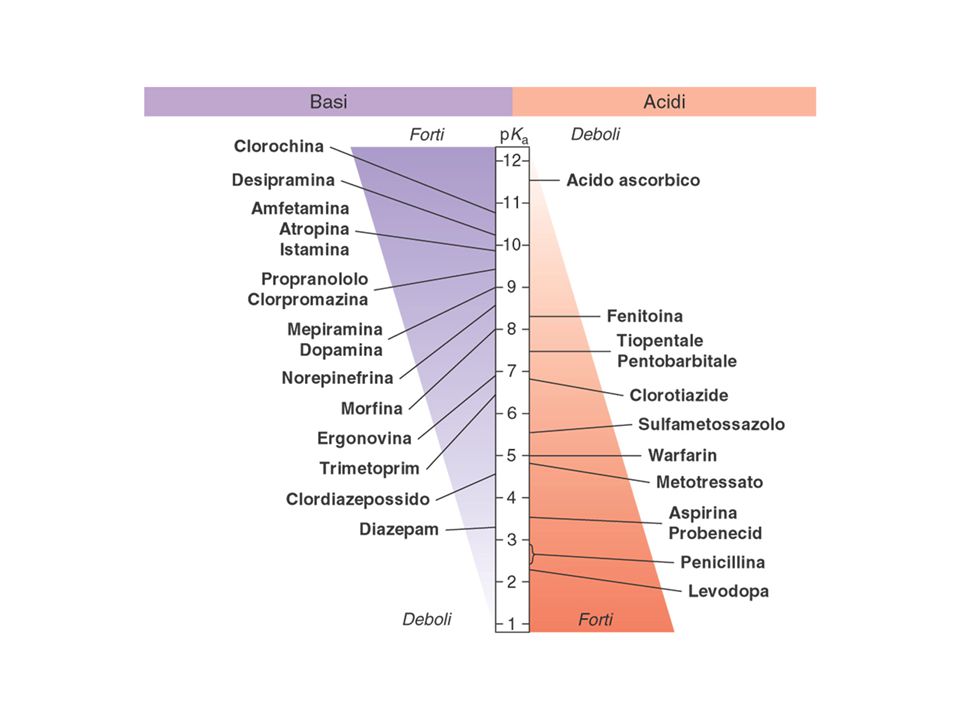
DIFFUSIONE PASSIVA Per diffondere attraverso una cellula, un F deve possedere un grado di idrofilia sufficiente a tenerlo in soluzione nei liquidi acquosi intra- ed extra-cellulari e un grado di lipofilia sufficiente a permettergli di attraversare la matrice lipidica delle membrane cellulari. La capacità di un farmaco di attraversare le membrane cellulari dipende dal suo coefficiente di ripartizione: CR=[F]o/[F]a CR ≈ 0 (F molto idrofilo => non diffonde attraverso le membrane) CR > 1 (F tendenzialmente lipofilo) Se CR è molto alto, il F non diffonde perché tende ad accumularsi nello spessore della matrice lipidica. CR dipende da: caratteristiche chimico-fisiche di una sostanza (conferiscono idrofilia i gruppi capaci di dare legami a idrogeno con H2O, es. -OH, -NH2, C=O, etc.) pH dell’ambiente (può modificare la carica di gruppi acidi o basici) (o=fase oleosa, a=fase acquosa)
 CR > 1 (F tendenzialmente lipofilo) Se CR è molto alto, il F non diffonde perché tende ad accumularsi nello spessore della matrice lipidica. CR dipende da: caratteristiche chimico-fisiche di una sostanza (conferiscono idrofilia i gruppi capaci di dare legami a idrogeno con H2O, es. -OH, -NH2, C=O, etc.) pH dell’ambiente (può modificare la carica di gruppi acidi o basici) (o=fase oleosa, a=fase acquosa)")
16
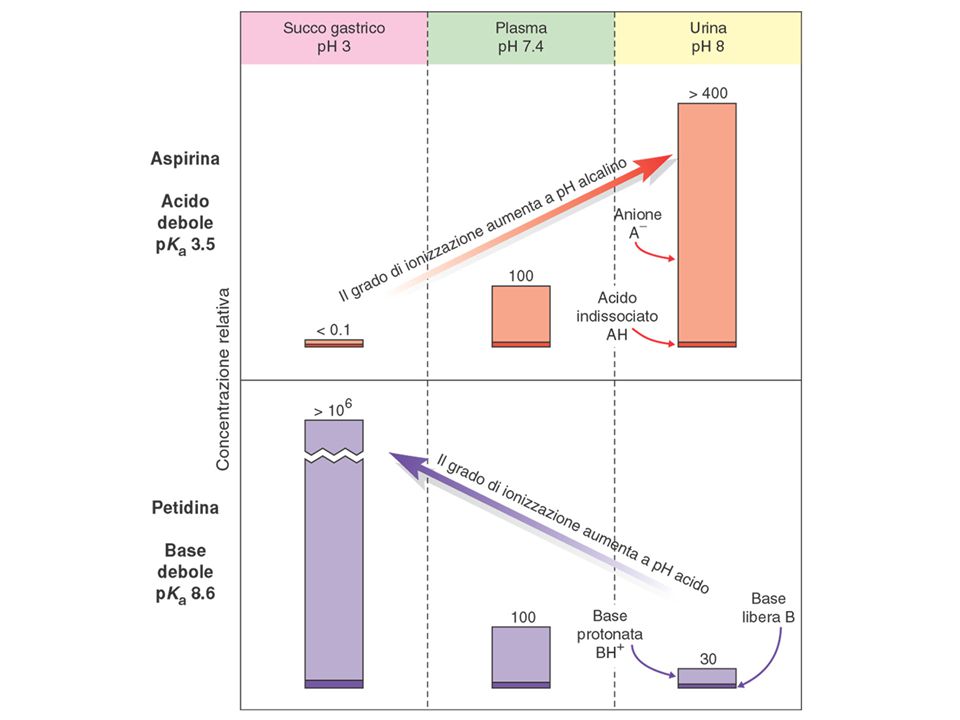
Ka=[A-] [H+]/[HA]; pKa= - log Ka => [A-]/[HA] = 10 (pH-pKa)
ASA (pKa = 3,5) Per le basi deboli il fenomeno è opposto => si accumulano sul lato più acido della membrana
![Ka=[A-] [H+]/[HA]; pKa= - log Ka => [A-]/[HA] = 10 (pH-pKa)](http://slideplayer.it/slide/2663495/9/images/16/Ka%3D%5BA-%5D+%5BH%2B%5D%2F%5BHA%5D%3B+pKa%3D+-+log+Ka+%3D%3E+%5BA-%5D%2F%5BHA%5D+%3D+10+%28pH-pKa%29.jpg "ASA (pKa = 3,5) Per le basi deboli il fenomeno è opposto => si accumulano sul lato più acido della membrana.")
20
LEGGE DI FICK Solo un F dotato di CR adeguato può diffondere attraverso le membrane cellulari. La diffusione tra due compartimenti separati da una membrana è governata dalla legge di Fick: Flusso molare = (C1 – C2) D x A / d Flusso molare = velocità (moli/sec) del passaggio di un soluto dal compartimento 1 al compartimento 2 C1 e C2 = concentrazioni del composto nei due compartimenti (quanto > è la differenza di conc fra i due compartimenti, tanto > è il flusso) D = coefficiente di diffusione (dipende dalle caratteristiche chimico-fisiche del solvente e del soluto; nel caso di diffusione attraverso membrane biologiche dipende principalmente dal CR del farmaco) A = area della membrana che separa i due compartimenti (es. intestino tenue) d = spessore della membrana (membrana plasmatica o numero di strati di cellule di un tessuto da superare) (per es. cute vs mucose).
 D x A / d. Flusso molare = velocità (moli/sec) del passaggio di un soluto dal compartimento 1 al compartimento 2. C1 e C2 = concentrazioni del composto nei due compartimenti (quanto > è la differenza di conc fra i due compartimenti, tanto > è il flusso) D = coefficiente di diffusione (dipende dalle caratteristiche chimico-fisiche del solvente e del soluto; nel caso di diffusione attraverso membrane biologiche dipende principalmente dal CR del farmaco) A = area della membrana che separa i due compartimenti (es. intestino tenue) d = spessore della membrana (membrana plasmatica o numero di strati di cellule di un tessuto da superare) (per es. cute vs mucose).")
22
PASSAGGIO DEI FARMACI ATTRAVERSO LE MEMBRANE CELLULARI
I processi di assorbimento, distribuzione, metabolismo ed eliminazione sono tutti influenzati dalla capacità del farmaco di attraversare le membrane cellulari. Per es., a seguito di somministrazione orale, il F deve attraversare l’epitelio gastro-intestinale, penetrare nel circolo linfatico/ematico e lasciarlo in un altro punto dell’organismo, eventualmente superando altre barriere (es. BEE). Intanto il F è sottoposto all’azione metabolica di enzimi circolanti, ma se è capace di penetrare all’interno delle cellule viene metabolizzato da enzimi intracellulari (es. a livello epatico) o addirittura può essere trasportato dalle cellule (es. azitromicina). Anche l’escrezione renale dipende dalla capacità del farmaco di essere riassorbito a livello dell’epitelio tubulare. Tre modalità di passaggio: Diffusione passiva Endo-esocitosi Trasporto proteina-mediato (carriers, simporti, antiporti e pompe)
. Intanto il F è sottoposto all’azione metabolica di enzimi circolanti, ma se è capace di penetrare all’interno delle cellule viene metabolizzato da enzimi intracellulari (es. a livello epatico) o addirittura può essere trasportato dalle cellule (es. azitromicina). Anche l’escrezione renale dipende dalla capacità del farmaco di essere riassorbito a livello dell’epitelio tubulare. Tre modalità di passaggio: Diffusione passiva. Endo-esocitosi. Trasporto proteina-mediato (carriers, simporti, antiporti e pompe)")
23
TRASPORTO ATTRAVERSO LE MEMBRANE
Consente il passaggio di farmaci il cui CR non ne permetterebbe la diffusione passiva. Si distinguono 4 processi: Trasporto dovuto a processi di endo-, pino- o transcitosi Trasporto attivo contro gradiente dovuto a proteine trasportatrici o scambiatrici Diffusione facilitata mediata da trasportatori o carriers Passaggio attraverso canali o pori

24
ENDOCITOSI: il F viene inglobato in vescicole che si formano per invaginazione della membrana (endocitosi) che possono poi scaricare il loro contenuto per esocitosi sul versante cellulare opposto (transcitosi: processo tipico dell’endotelio capillare). TRASPORTO ATTIVO: scambiatori/cotrasportatori (antiporti e simporti) o pompe garantiscono il trasporto del F contro gradiente elettrochimico, grazie all’accoppiamento ad un altro processo in grado di fornire energia. Il flusso è solo parzialmente influenzato dalla differenza di conc del F; il trasporto è saturabile (il flusso max dipende dalla densità del trasportatore e dalla sua velocità di trasporto). DIFFUSIONE FACILITATA: avviene attraverso i carriers: proteine di membrana capaci di esporre siti specifici alternativamente sulle due superfici della membrana. Il legame di ioni o molecole cariche a questi siti, maschera i loro gruppi polari, consentendone l’attraversamento di membrana e favorendone il rilascio dall’altra parte. I carriers sono generalmente molto selettivi. Il flusso del F segue le leggi della diffusione passiva (dipende dal gradiente di conc), ma oltre una conc critica diventa saturabile. PASSAGGIO ATTRAVERSO CANALI O PORI: consente il passaggio di ioni e molecole idrofile o cariche. I canali sono costituiti da proteine transmembranarie che formano al loro interno un poro idrofilo (aa carichi). Il passaggio del farmaco segue le leggi della diffusione e avviene secondo gradiente elettrochimico. A seconda dei gruppi carichi esposti nel lume, il poro può essere selettivo (x ioni specifici, cationi o anioni, dimensione ione, etc).
 o pompe garantiscono il trasporto del F contro gradiente elettrochimico, grazie all’accoppiamento ad un altro processo in grado di fornire energia. Il flusso è solo parzialmente influenzato dalla differenza di conc del F; il trasporto è saturabile (il flusso max dipende dalla densità del trasportatore e dalla sua velocità di trasporto). DIFFUSIONE FACILITATA: avviene attraverso i carriers: proteine di membrana capaci di esporre siti specifici alternativamente sulle due superfici della membrana. Il legame di ioni o molecole cariche a questi siti, maschera i loro gruppi polari, consentendone l’attraversamento di membrana e favorendone il rilascio dall’altra parte. I carriers sono generalmente molto selettivi. Il flusso del F segue le leggi della diffusione passiva (dipende dal gradiente di conc), ma oltre una conc critica diventa saturabile. PASSAGGIO ATTRAVERSO CANALI O PORI: consente il passaggio di ioni e molecole idrofile o cariche. I canali sono costituiti da proteine transmembranarie che formano al loro interno un poro idrofilo (aa carichi). Il passaggio del farmaco segue le leggi della diffusione e avviene secondo gradiente elettrochimico. A seconda dei gruppi carichi esposti nel lume, il poro può essere selettivo (x ioni specifici, cationi o anioni, dimensione ione, etc).")
25
CARATTERISTICHE DELLE PRINCIPALI BARRIERE CELLULARI
La velocità del passaggio di un F da un compartimento all’altro è determinata da: Legge di Fick Densità dei sistemi di trasporto Flusso specifico (litri di plasma per grammo di tessuto per minuto; CNS>muscolo)) Caratteristiche strutturali e funzionali della barriera Le caratteristiche strutturali e funzionali delle varie barriere biologiche influenzano non solo l’efficienza e la velocità di passaggio di un farmaco, ma anche il grado di selettività della barriera in funzione delle caratteristiche fisico-chimiche dei diversi farmaci e della presenza di meccanismi selettivi di traslocazione. ENDOTELIO CAPILLARE BARRIERA EMATO-ENCEFALICA (BEE) BARRIERA PLACENTARE
) Caratteristiche strutturali e funzionali della barriera. Le caratteristiche strutturali e funzionali delle varie barriere biologiche influenzano non solo l’efficienza e la velocità di passaggio di un farmaco, ma anche il grado di selettività della barriera in funzione delle caratteristiche fisico-chimiche dei diversi farmaci e della presenza di meccanismi selettivi di traslocazione. ENDOTELIO CAPILLARE. BARRIERA EMATO-ENCEFALICA (BEE) BARRIERA PLACENTARE.")
26
ENDOTELIO CAPILLARE E’ una barriera molto labile: spessore limitato, alta attività di endo-esocitosi, presenza di fenestrae e pori membranari. La ricchezza dell’irrorazione capillare (da 50 capillari/mm2 nella cute a 2000 nel miocardio) rende disponibile un’estesissima superficie di scambio => rapida diffusione sangue-liquidi interstiziali Molecole idrofile e polari con PM<60 KDa: passano attraverso i pori (Ø ~ 10 nm) localizzati fra cellule endoteliali adiacenti o a livello delle fenestrae. Molecole liposolubili (inclusi i gas): diffondono attraverso le membrane e la velocità di diffusione dipende dal CR. NB: - solo le molecole libere in soluzione sono capaci di attraversare la barriera endoteliale (aggregazione o legame alla proteine plasmatiche limitano il passaggio) alterazioni morfo-funzionali dell’endotelio capillare (es. infiammazione) possono influenzare la velocità di diffusione variazioni della perfusione tissutale (es. costrizione arteriolare) possono limitare la diffusione agli spazi interstiziali
 rende disponibile un’estesissima superficie di scambio => rapida diffusione sangue-liquidi interstiziali. Molecole idrofile e polari con PM<60 KDa: passano attraverso i pori (Ø ~ 10 nm) localizzati fra cellule endoteliali adiacenti o a livello delle fenestrae. Molecole liposolubili (inclusi i gas): diffondono attraverso le membrane e la velocità di diffusione dipende dal CR. NB: - solo le molecole libere in soluzione sono capaci di attraversare la barriera endoteliale (aggregazione o legame alla proteine plasmatiche limitano il passaggio) alterazioni morfo-funzionali dell’endotelio capillare (es. infiammazione) possono influenzare la velocità di diffusione. variazioni della perfusione tissutale (es. costrizione arteriolare) possono limitare la diffusione agli spazi interstiziali.")
27
La permeabilità capillare è diversa nei vari distretti:

28
LA BARRIERA EMATO-ENCEFALICA (BEE)
I F idrofili passano solo se la BEE è compromessa (es. aterosclerosi nell’anziano, meningite, stati febbrili elevati specialmente nei bambini) o mediante trasportatori (es. L-DOPA) o endocitosi mediata da recettore (es. insulina, fattori di crescita, transferrina).
 o mediante trasportatori (es. L-DOPA) o endocitosi mediata da recettore (es. insulina, fattori di crescita, transferrina).")
29
LA BARRIERA PLACENTARE
La placenta è caratterizzata dalla presenza di seni ematici materni nei quali si spingono i villi irrorati dalla circolazione capillare fetale. Il sangue fetale è separato dal sangue materno dal sincizio placentare, dall’interstizio villare e dalle cellule endoteliali dei capillari villari. Il sincizio placentare costituisce un filtro molecolare nel quale sono attivi i diversi meccanismi di passaggio: diffusione passiva, diffusione facilitata (glucosio), trasporto attivo (aa, Calcio, fosfato), endocitosi mediata da recettore (transferrina, IgG e altre proteine materne). Lo spessore della BP è di 25mm all’inizio della gestazione e di 2mm al termine. Poiché il sangue materno fluisce molto lentamente, la probabilità di un farmaco di attraversare il sincizio e arrivare al feto è elevata. Il passaggio di F dalla madre al feto è trascurabile solo per F idrofili di grosse dimensioni, presenti in concentrazione relativamente bassa ed eliminati rapidamente dal circolo materno. Il legame dei F alle proteine del feto, rallentano il raggiungimento dell’equilibrio e rendono il Vd apparente del feto molto elevato (=> accumulo).
, trasporto attivo (aa, Calcio, fosfato), endocitosi mediata da recettore (transferrina, IgG e altre proteine materne). Lo spessore della BP è di 25mm all’inizio della gestazione e di 2mm al termine. Poiché il sangue materno fluisce molto lentamente, la probabilità di un farmaco di attraversare il sincizio e arrivare al feto è elevata. Il passaggio di F dalla madre al feto è trascurabile solo per F idrofili di grosse dimensioni, presenti in concentrazione relativamente bassa ed eliminati rapidamente dal circolo materno. Il legame dei F alle proteine del feto, rallentano il raggiungimento dell’equilibrio e rendono il Vd apparente del feto molto elevato (=> accumulo).")
Presentazioni simili


















, specializzato per la distribuzione di: gas.>")



