Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Nicoletta Fortunati SCDU Endocrinologia Oncologica
CARCINOMA della MAMMELLA: BASI MOLECOLARI ed IMPLICAZIONI PROGNOSTICHE e TERAPEUTICHE Nicoletta Fortunati SCDU Endocrinologia Oncologica 5 Ottobre 2011 Corso di aggiornamento in Endocrinologia Clinica Endocrinologia oncologica “Hot topics” in clinica, diagnosi e terapia

2
TARGETED THERAPY TARGET

3
CARCINOMA MAMMARIO SOTTOTIPI
LUMINAL A: ERa o PgR positivi, HER2 negativo LUMINAL B: ERa o PgR positivi, HER2 positivo HER2 POSITIVO: overespressione di HER2 BASAL: ERa e PgR negativi, HER2 negativo; (TRIPLO NEGATIVO)
")
4
SHRs:domains funzionali e omologie
Da: Ahmad N & Kumar R, Cancer Lett 300: 1-9, 2011

5
ERa & ERb Da: Leitman Dc et al. Curr Op Pharmacol 10: , 2010

6
ERa & ERb DIFFERENZE Prodotti di 2 differenti geni (ERa, locus 11q13.1; ERb, locus 14q23.2 ) LBD presenta una dimensione diversa AF-1 e AF-2 sono localizzati nelle regione meno conservata ERa richiede sempre la presenza del ligando per avviare la regolazione genica mentre ERb è attivo anche nella forma non legata Regolano sets di geni differenti sia perché si legano a regioni diverse del DNA (diverse dal tradizionale ERE) sia perché reclutano diversi sets di coregolatori
 sia perché reclutano diversi sets di coregolatori.")
7
Meccanismo Classico dell’Azione degli SHRs
Da: Ahmad N & Kumar R, Cancer Lett 300: 1-9, 2011

8
SHRs SHRs sono fattori di trascrizione ligando-attivati che comprendono i recettori per glucocorticoidi (GR), estrogeni (ER), progesterone (PR), androgeni (AR), e mineralocorticoidi (MR) SHRs sono costituiti da almeno 3 domains funzionali principali, che presentano diverse funzioni e diversi gradi di omologia: N-terminale (NTD)-, DNA binding domain (DBD-) il più conservato, e ligand binding (LBD)-domain C-terminale. Il modello d’azione classico prevede che gli SHRs non legati si trovino nel compartimento citoplasmatico associati con varie heat shock e altre protein chaperone; quando si lega il ligando, il recettore va incontro a rearrangiamenti conformazionali e transloca nel nucleo dove si lega a specifiche sequenze del DNA conosciute come hormone response elements (HREs) e si assembla con le proteine partner coregolatorie.
, estrogeni (ER), progesterone (PR), androgeni (AR), e mineralocorticoidi (MR) SHRs sono costituiti da almeno 3 domains funzionali principali, che presentano diverse funzioni e diversi gradi di omologia: N-terminale (NTD)-, DNA binding domain (DBD-) il più conservato, e ligand binding (LBD)-domain C-terminale. Il modello d’azione classico prevede che gli SHRs non legati si trovino nel compartimento citoplasmatico associati con varie heat shock e altre protein chaperone; quando si lega il ligando, il recettore va incontro a rearrangiamenti conformazionali e transloca nel nucleo dove si lega a specifiche sequenze del DNA conosciute come hormone response elements (HREs) e si assembla con le proteine partner coregolatorie.")
9
COREGOLATORI [COATTIVATORI & COREPRESSORI]
Coattivatori e corepressori sono presenti nello stesso complesso molecolare che controlla il meccanismo trascrizionale regolato dagli SHRs e che ha come ultimo effetto l’espressione di specifici geni targets Questi complessi multi-molecolari agiscono stabilizzando la macchina trascrizionale basale e/o rimodellando la cromatina, funzione nella quale sono implicate numerose attività enzimatiche come le istone acetyltransferasi e deacetylasi La disregolazione del complesso SHR/coregolatori è coinvolta nello sviluppo di patologie quali le Sindromi da Resistenza Ormonale e le Neoplasie Ormono-dipendenti. I sets di coregolatori utilizzati cambia da tessuto a tessuto
![COREGOLATORI [COATTIVATORI & COREPRESSORI]](http://slideplayer.it/slide/1414751/8/images/9/COREGOLATORI+%5BCOATTIVATORI+%26+COREPRESSORI%5D.jpg "Coattivatori e corepressori sono presenti nello stesso complesso molecolare che controlla il meccanismo trascrizionale regolato dagli SHRs e che ha come ultimo effetto l’espressione di specifici geni targets. Questi complessi multi-molecolari agiscono stabilizzando la macchina trascrizionale basale e/o rimodellando la cromatina, funzione nella quale sono implicate numerose attività enzimatiche come le istone acetyltransferasi e deacetylasi. La disregolazione del complesso SHR/coregolatori è coinvolta nello sviluppo di patologie quali le Sindromi da Resistenza Ormonale e le Neoplasie Ormono-dipendenti. I sets di coregolatori utilizzati cambia da tessuto a tessuto.")
10
FARMACOLOGIA degli SHRs
Scoperta di meccanismi di specificità del tessuto target che moduli l’azione agonista o antagonista del farmaco Sviluppo di modulatori degli SHRs ad attività tessuto-selettiva La terapia ormonale è fondamentale nel trattamento di alcune neoplasie (e.g. mammella e prostata) ma la durata e le dosi dei modulatori degli SHRs sono ancora in via di definizione anche alla luce dello sviluppo della resistenza a tali farmaci che si sviluppa nel tempo questo è ritenuto un problema clinico maggiore che richiede lo sviluppo di nuovi agenti farmacologici in grado di superare la resistenza ai modulatori degli SHRs
 ma la durata e le dosi dei modulatori degli SHRs sono ancora in via di definizione anche alla luce dello sviluppo della resistenza a tali farmaci che si sviluppa nel tempo questo è ritenuto un problema clinico maggiore che richiede lo sviluppo di nuovi agenti farmacologici in grado di superare la resistenza ai modulatori degli SHRs.")
11
SERMS: TAMOXIFENE

12
BASI MOLECOLARI della RESISTENZA alla TERAPIA ENDOCRINA
Resistenza de novo o acquisita Perdita di ERa (soppressione espressione del recettore e/o selezione clonale di cellule ER-negative) Aumentata attività delle vie indotte da EGFR e/o IGFR con inibizione della funzionalità di ERa Ipersensibilità agli estrogeni dopo deprivazione
 Aumentata attività delle vie indotte da EGFR e/o IGFR con inibizione della funzionalità di ERa. Ipersensibilità agli estrogeni dopo deprivazione.")
13
MECCANISMI “NON-CLASSICI” dell’AZIONE ESTROGENICA
Signalling ERa-mediato ligando-indipendente (attivazione di EGF-R, IGF-R, GProtein coupled-R, che inducono kinasi e fosfatasi e modificano lo stato fosforilativo di ERa) Effetti rapidi, “non-genomici”,mediati da un recettore membrane-associato Signalling ERE-indipendente (trascrizione genica regolata da interazione proteina-proteina tra ERa e altri fattori trascrizionali come AP-1, Sp1, NF-kB)
 Effetti rapidi, non-genomici ,mediati da un recettore membrane-associato. Signalling ERE-indipendente (trascrizione genica regolata da interazione proteina-proteina tra ERa e altri fattori trascrizionali come AP-1, Sp1, NF-kB)")
14
Da: Barone I et al. Clin Cancer Res 2010;16:2702-2708

15
Da: Barone I et al. Clin Cancer Res 2010;16:2702-2708

16
KINASI & ERa Il tipo di kinasi che viene attivata determina risposte differenti al ligando Erk1/2 → S118; Akt → S167 → resistenza al tamoxifene; → attivazione ligando-indipendente PKA e PAK-1 → S305 → alterazione della sensibilità agli estrogeni e al tamoxifene

17
Da: Barone I et al. Clin Cancer Res 2010;16:2702-2708

18
HINGE REGION Domain multifunzionale che lega numerosi coregolatori e partecipa al legame di ERa al DNA Acetilazione di K266 e K268 → induce il legame al DNA e l’attivazione ligando indipendente Acetilazione di K302 e K303 → inibisce l’attivazione di ERa Fosforilazioni delle Ser ed Acetilazione delle Lys sono tra di loro collegate (S305 → K302/303)
")
19
Da: Barone I et al. Clin Cancer Res 2010;16:2702-2708

20
VARIANTI DI ERa Sono note numerose varianti di splicing di ERa che sono co-espresse con il recettore wild type con effetto dominante positivo o negativo; in questo caso conferiscono alle cellule di Carcinoma Mammario fenotipo ormono-indipendente ERa D3 → dominante negativo; sopprime attività trascrizionale estrogeno-indotta; ↓ crescita cellulare ancoraggio-dipendente; ↓ formazione di colonie in soft agar; ↓potenziale invasivo in vivo hERa-46 → splicing alternativo di esone 1 che determina la mancanza di 173 aa in AF-1; co-localizza sulle membrane con ERa full-length; inibitore competitivo di ERa; promuove attività genomica in cellule ER-negative hERa-36 → trascrizione da promoter alternativo del gene di ERa66; mancano entrambi AF-1 e AF-2; conservati DBD e parte di HBD; possiede un domain extra di 27 aa; localizzata a livello di membrana dove trasduce la cascata di segnale “non-genomico” mediato da estrogeni ed antiestrogeni; inibisce effetti genomici di ERa; overexpression in carcinomi si accompagna a scarsa prognosi e resistenza al tamoxifene

21
Da: Barone I et al. Clin Cancer Res 2010;16:2702-2708

22
MUTAZIONI SPONTANEE di ERa
Y537N → elimina un residuo di Tyr carbossi-terminale sito critico di fosforilazione → recettore costituzionalmente attivo (presente in alcuni carcinomi mammari metastatici) K303R → sostituisce una Lys con Arg nel sito 303 della Hinge che causa una maggiore fosforilazione da parte di PKA e Akt → ipersensibilità agli estrogeni, ridotta sensibilità al tamoxifene, resistenza all’anastrazolo (presente in circa 1/3 delle iperplasie mammarie premaligne)
 K303R → sostituisce una Lys con Arg nel sito 303 della Hinge che causa una maggiore fosforilazione da parte di PKA e Akt → ipersensibilità agli estrogeni, ridotta sensibilità al tamoxifene, resistenza all’anastrazolo (presente in circa 1/3 delle iperplasie mammarie premaligne)")
23
INIBITORI AROMATASI INIBITORI AROMATASI

24
INIBITORI AROMATASI

25
INIBITORI AROMATASI

26
INIBITORI AROMATASI EXEMESTANO ANASTRAZOLO

27
Da: Johnston S R Clin Cancer Res 2010;16:1979-1987

28
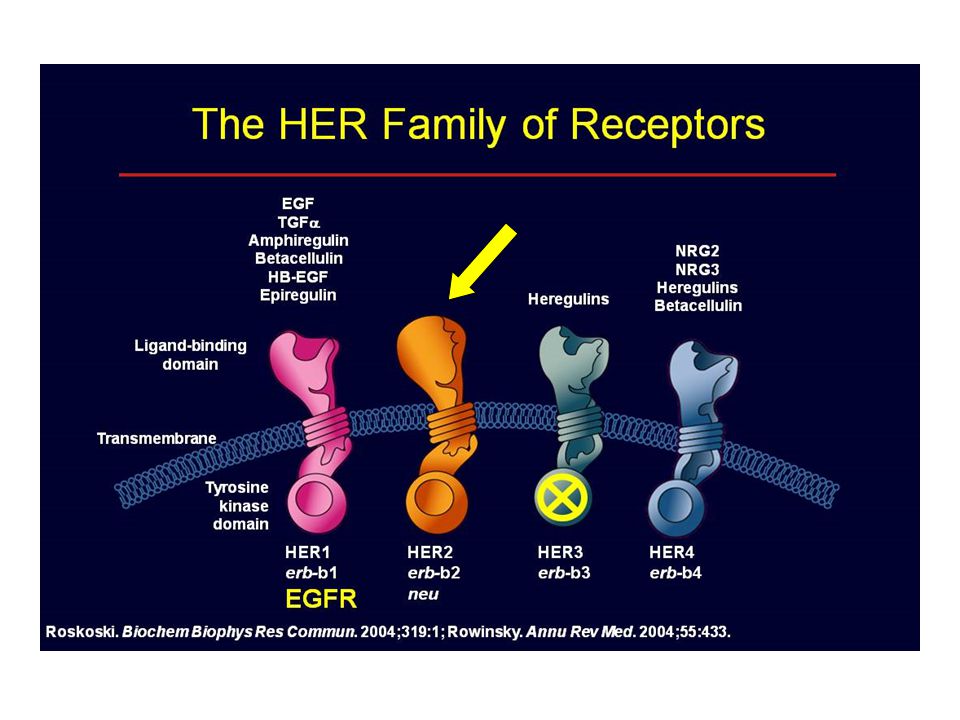
HER2 Appartiene alla famiglia dei recettori per Epidermal Growth Factor (TK) 4 recettori Tirosino-Kinasi (TK) di membrana: HER1, HER2, HER3, HER4 HER2 è overespresso in 20-25% dei carcinomi mammari; dovuta ad amplificazione del gene HER2; associata a fenotipo aggressivo e prognosi peggiore Non ha un ligando proprio ma si attiva dopo etero dimerizzazione con un altro membro della famiglia

30
HERs & LIGANDI Da: Gutierrez C, Schiff R, Arch Pathol Lab Med 2011

31
HER1, HER2, HER3, HER4

32
HER: SIGNALLING PATHWAYS
Da Linden H M et al. Clin Cancer Res 2006; 12:

33
HER-2 SCORING - IMMUNOISTOCHIMICA
1+ 2+ 3+ Da: Gutierrez C, Schiff R, Arch Pathol Lab Med 2011

34
HER-2 GENE AMPLIFICATION
Da: Gutierrez C, Schiff R, Arch Pathol Lab Med 2011

35
HER2 OVEREXPRESSION Da: Gutierrez C, Schiff R, Arch Pathol Lab Med 2011

36
HER-2 TARGETED THERAPY Da: Gutierrez C, Schiff R, Arch Pathol Lab Med 2011

37
TRASTUZUMAB

38
LAPATINIB

39
PERTUZUMAB

40
NERATINIB da Alvarez, R. H. et al. J Clin Oncol; 28:

41
INIBITORI di mTOR

42
ANGIOGENESI

43
VEGFR

44
INIBITORI VEGF e VEGFR

45
BEVACIZUMAB

46
VGFR/TK-INIBITORI SUNITINIB SORAFENIB VATALANIB MOTESANIB

47
PARP (Poly Adenosine-diphosphate Ribose)
")
48
BRCA1 e BRCA2 Cellule con mutazioni loss-of-function di BRCA possono perdere l’allele wild type alterazioni di BRCA1 e BRCA2 sensibilizzano le cellule all’azione di PARP → instabilità cromosomica → aberrazioni genetiche → carcinogenesi Solo le cellule mutazioni BRCA1 e BRCA2 sono sensibili agli inibitori di PARP

49
INIBITORI di PARP

50
FARMACI TARGET di PATHWAYS che interagiscono con ERα in CARCINOMI MAMMARI ERα-POSITIVI: CLINICAL TRIALS Farmaco/Combinazione Pathway Target(s) Stato Malattia Fase PD * MEK (MAPK pathway) Caricinoma mammario, colon melanoma; avanzato I-II Bevacizumab + sorafenib tosylate† RAF (MAPK pathway) Tumori solidi refrattari, metastatici, non operabili I Paclitaxel and RAD001 followed by FEC (chemotherapy)‡§ mTOR Carcinoma mammario Triplo Negativo II Ritonavir (preoperative)∥ Akt Carcinoma mammario di nuova diagnosi GSK ¶ Tumori solidi/linfomi non responsivi ad altre terapie GDC bevacizumab + paclitaxel** PI3K Carcinoma mammario metatstico o con recidive locali Ib BGT226§ Tumori solidi avanzati (anche Carcinoma mammario) BEZ235§ Temsirolimus††† XL147+ XL647‡‡ Tumori solide (anche Carcinoma mammario)
 Stato Malattia. Fase. PD * MEK (MAPK pathway) Caricinoma mammario, colon melanoma; avanzato. I-II. Bevacizumab + sorafenib tosylate† RAF (MAPK pathway) Tumori solidi refrattari, metastatici, non operabili. I. Paclitaxel and RAD001 followed by FEC (chemotherapy)‡§ mTOR. Carcinoma mammario Triplo Negativo. II. Ritonavir (preoperative)∥ Akt. Carcinoma mammario di nuova diagnosi. GSK ¶ Tumori solidi/linfomi non responsivi ad altre terapie. GDC bevacizumab + paclitaxel** PI3K. Carcinoma mammario metatstico o con recidive locali. Ib. BGT226§ Tumori solidi avanzati (anche Carcinoma mammario) BEZ235§ Temsirolimus††† XL147+ XL647‡‡ Tumori solide (anche Carcinoma mammario)")
51
TARGET TARGETED THERAPY

52
GRAZIE

Presentazioni simili





, L Garagnani (2), L Schirosi (3), C De Gaetani (4), A Maiorana.>")


>")










