Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
ANEMIE EMOLITICHE

2
1% della massa eritrocitaria distrutta ogni giorno negli organi emocateretici
Sopravvivenza normale degli eritrociti in circolo circa 120 giorni Anemia può mancare se iperemolisi è compensata dal midollo osseo L’ittero può mancare se il fegato elabora la quantità di bilirubina indiretta prodotta in eccesso

3
Le Anemie Emolitiche caratterizzate dalla Anemie distruzione
e quindi riduzione della vita media dei globuli rossi

4
Definizione di anemia emolitica
Anemia dovuta alla diminuzione della durata di vita del globulo rosso non adeguatamente compensata dall’aumento di produzione midollare.

5
Anemie emolitiche Reticolocitosi Iperplasia eritroblastica
SEGNI DI IPERFUNZIONE COMPENSATORIA ERITRONE Reticolocitosi Iperplasia eritroblastica Aumento del turnover del ferro plasmatico

6
Anemie emolitiche SEGNI DI EMOLISI Aumento della bilirubina indiretta
Aumento urobilinuria e bilinogeno fecale Riduzione vita media emazie (Emoglobinemia,emoglobinuria)
")
7
Caratteri anemie emolitiche
Emoglobina diminuita(Hb) Reticolociti aumentati Aptoglobina diminuita o assente LDH aumenata Bilirubina aumentata Iperplasia eritroide midollare +/- associata ad aumento % dei linfociti
 Reticolociti aumentati. Aptoglobina diminuita o assente. LDH aumenata. Bilirubina aumentata. Iperplasia eritroide midollare +/- associata ad aumento % dei linfociti.")
8
Anemia Emolitica Anemia normocromica normocitica. O macrocitica
Reticolocitosi. Riduzione dell’aptoglobina. Iperbilirubinemia indiretta, aumento dell’LDH. Anemia Emolitica

9
Diagnosi di anemia emolitica
Anamnesi Età, sesso, familiarità, luogo di nascita. Precedenti trasfusionali. Tempo di comparsa, durata, tipo di sintomi. Precedenti di ittero, subittero e/o calcolosi biliare. Esposizione a farmaci, tossici. Presenza di altre patologie. Precedenti crisi dolorose o vaso-occlusive.

10
Diagnosi di anemia emolitica
Indagini di I livello Esame emocromocitometrico. Conta reticolocitaria. Esame microscopico dello striscio di sangue periferico. Bilirubina totale e frazionata. LDH. Sideremia e ferritina sierica, sTfR. Funzionalità renale. Esame urine: urobilinuria, emoglobinuria, emosiderinuria. Studio coagulazione. Aptoglobina.

11
Indagini di II livello per anemia emolitica (diagnosi differenziale)
Elettroforesi dell’emoglobina. Resistenza osmotica eritrocitaria, test autoemolisi Indagini immunoematologiche. Test di Ham e citofluorimetria a flusso Dosaggio degli enzimi eritrocitari. Biologia molecolare.

12
Anemia Emolitica Extraglobulari Miste Extravascolare Cronica
1) EMOLISI Intravascolare Extravascolare 2) PRESENTAZIONE CLINICA Acuta Cronica 3) CAUSE Intraglobulari Extraglobulari Miste
 EMOLISI. Intravascolare. Extravascolare. 2) PRESENTAZIONE CLINICA. Acuta. Cronica. 3) CAUSE. Intraglobulari. Extraglobulari. Miste.")
13
Anemia da aumentata distruzione Emolisi Intraglobulare Extraglobulare
ANEMIA EMOLITICA Anemia da aumentata distruzione Emolisi Intraglobulare Extraglobulare

14
Meccanismi dell’Emocateresi
Emolisi Extravascolare ( SRE: milza, fegato…) Complessi Hb+Apt Fegato Intravascolare Hb libera in circolo Eme+ albumina Filtrato glomerulare Rene Riassorbimento tubulare
 Complessi. Hb+Apt. Fegato. Intravascolare Hb libera. in circolo. Eme+ albumina. Filtrato. glomerulare. Rene. Riassorbimento. tubulare.")
15
Emolisi Extraglobulare Fegato, Milza
Urine ipercromoche (iperurobilinuria) Feci scure (aumento bilinogeno fecale) Intraglobulare Nel sistema circolatorio Emoglobinemia Emoglobinuria
 Feci scure. (aumento bilinogeno fecale) Intraglobulare. Nel sistema circolatorio. Emoglobinemia. Emoglobinuria.")
16
Le Anemie Emolitiche CLINICA
FORME ACUTE pallore ingravescente sudorazione fredda Febbre elevata, brivido, Cefalea dolori lombari subittero FORME CRONICHE pallore modico, ittero di gravità variabile splenomegalia/epato- megalia variabili colelitiasi(calcoli di biliru- binato) disturbi trofici:ulcere agli arti inferiori
 disturbi trofici:ulcere agli. arti inferiori.")
17
Le Anemie Emolitiche CLINICA
FORME ACUTE Emolisi prevalentemente intravascolare: Saturazione dell’aptoglobina Emoglobinemia (siero laccato) Emoglobinuria(urine rosso scuro o nere) Talora extravacolare (deficit G-6-PDH) FORME CRONICHE Emolisi prevalentemente extravascolare (macrofagi, SRE) Talora intravascolare (protesi valvolari meccaniche, EPN)
 Emoglobinuria(urine rosso. scuro o nere) Talora extravacolare. (deficit G-6-PDH) FORME CRONICHE. Emolisi prevalentemente. extravascolare. (macrofagi, SRE) Talora intravascolare. (protesi valvolari. meccaniche, EPN)")
18
Anemie emolitiche clinica
Crisi iperemolitiche acute Crisi aplastiche

19
CLASSIFICAZIONE DELLE ANEMIE EMOLITICHE
A. EMOLITICHE ACQUISITE A. EMOLITICHE CONGENITE ANEMIE EMOLITICHE ANTICORPO MEDIATE (caldi, freddi, bifasici) DA DIFETTO ENZIMATICO DA DIFETTO DI MEMBRANA EMOLISI MECCANICA EPN Deficit G6PD Deficit PK Sferocitosi ereditaria Ellissocitosi ereditaria - MEN - AEA - Farmaci - S. di Moschowitz - S. uremicoemolit. - Protesi Meccanica
 DA DIFETTO ENZIMATICO. DA DIFETTO DI MEMBRANA. EMOLISI MECCANICA. EPN. Deficit G6PD. Deficit PK. Sferocitosi ereditaria. Ellissocitosi ereditaria. - MEN. - AEA. - Farmaci. - S. di Moschowitz. - S. uremicoemolit. - Protesi Meccanica.")
20
Le Anemie Emolitiche Forme acquisite Forme congenite
immuni ( autoimmuni, isoimmuni, da farmaci) microangiopatiche da ipersplenismo Secondarie a malattie epatiche o renali Varie:infezioni, sostanze chimiche, tossiche EPN Forme congenite difetto di membrana enzimopenia emoglobinopatia
 microangiopatiche. da ipersplenismo. Secondarie a malattie epatiche o renali. Varie:infezioni, sostanze chimiche, tossiche. EPN. Forme congenite. difetto di membrana. enzimopenia. emoglobinopatia.")
21
Le Anemie Emolitiche -Sferocitosi -EPN -Malattie
Cause Cause Cause Intrinseche Miste Estrinseche -Sferocitosi EPN Malattie -Ellissocitosi emotiche -Deficit piruvato Carenza di G-6-PD -Microangio- kinasi patie -Hb instabili Cause mec- Hb S, C caniche - Farmaci

22
MECCANISMO EMOLISI Sferocitosi Cause meccaniche G6PD PK Autoimmnunità
INTRAGLOBULARE Sferocitosi G6PD PK EXTRAGLOBULARE Cause meccaniche Autoimmnunità

23
ANEMIE EMOLITICHE INTRAGLOBULARI

24
Anemie emolitiche intraglobulari
Le anemie emolitiche intraglobulari comprendono sia forme congenite sia forme acquisite Le forme congenite sono : Sferocitosi ereditaria (HS) Ellissocitosi ereditaria (HE) il deficit della glucosio 6 fosfato deidrogenasi il deficit di piruvato chinasi (PK) La forma acquisita è la : emoglobinuria parossistica notturna (EPN)
 Ellissocitosi ereditaria (HE) il deficit della glucosio 6 fosfato deidrogenasi. il deficit di piruvato chinasi (PK) La forma acquisita è la : emoglobinuria parossistica notturna (EPN)")
25
SFEROCITOSI

26
Sferositosi Ereditaria
Malattia CONGENITA a trasmissione AUTOSOMICA DOMINANTE , più raramente recessiva, dovuta a DIFETTO DELLA MEMBRANA ERITROCITARIA E’ l’anemia emolitica PIU’ FREQUENTE nel Nord Europa ( 1 soggetto ogni 5000)
")
27
Sferocitosi ereditaria (HS)
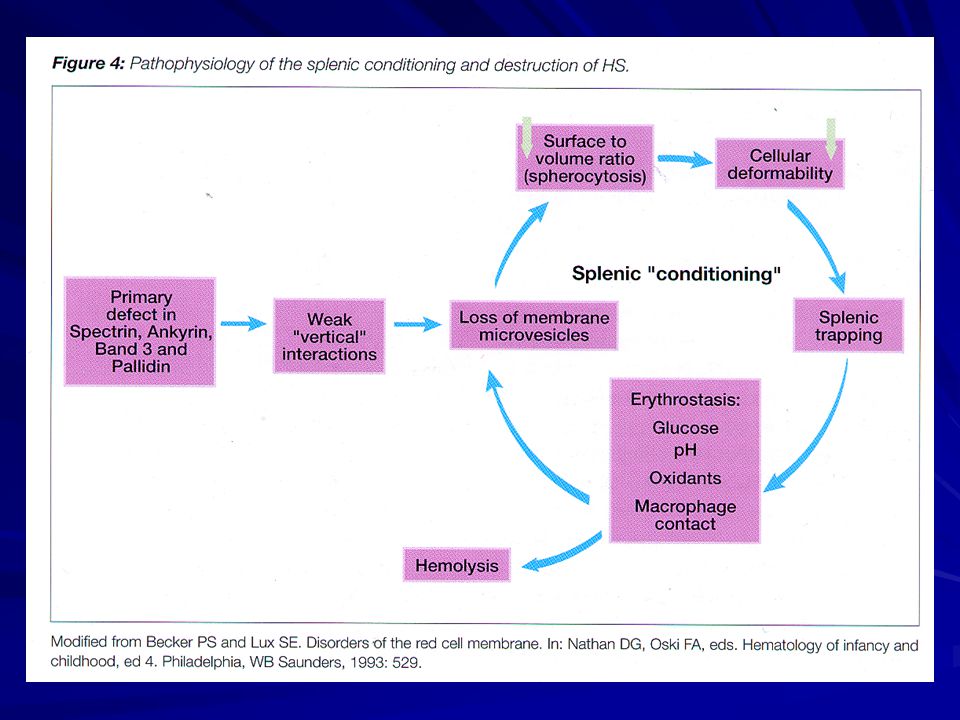
Anemia emolitica congenita a trasmissione autosomica, prevalentemente, dominante a penetranza variabile. L’alterazione coinvolge una proteina della membrana eritrocitaria,per deficit di sintesi o di struttura, prevalentemente, della α-spectrina e/o dell’ anchirina o della proteina della banda 3. Tali deficit producono una instabilità delle membrane lipidiche dello strato esterno eritrocitario, con conseguente formazione di microvescicole. Queste cellule sono osmoticamente fragili e sono sequestrate a livello splenico. La α-spectrina è generalmente sintetizzata in eccesso rispetto alla ß-spectrina, pertanto i soggetti con un solo allele inattivo sono asintomatici.

28
Struttura della membrana eritrocitaria

29
Sferositosi Ereditaria
PATOGENESI presenti difetti primitivi dei componenti del citoscheletro: (carenza di SPECTRINA primitiva o secondaria a difetto di anchirina, banda 3, proteina 4.2) con conseguente alterata coesione tra citoscheletro e strato lipidico
 con conseguente alterata coesione tra citoscheletro e strato lipidico.")
30
SCARSAMENTE DEFORMABILI , SONO TRATTENUTI E DANNEGGIATI DALLA MILZA.
riduzione della superficie eritrocitariamicrosferocitosi ed alterazione del trasporto cationico (pompa del sodio)accumulo di sodioimbibizione. GLI SFEROCITI , SCARSAMENTE DEFORMABILI , SONO TRATTENUTI E DANNEGGIATI DALLA MILZA.
accumulo di sodioimbibizione. GLI SFEROCITI , SCARSAMENTE DEFORMABILI , SONO TRATTENUTI E DANNEGGIATI. DALLA MILZA.")
32
H S : clinica Il quadro clinico è molto vario, essendo collegato al difetto molecolare, ed oscilla tra condizioni asintomatiche e condizioni di gravi anemie trasfusione dipendente. FORMA NEONATALE FORMA ADULTA Il quadro clinico è caratterizzato: da anemia più o meno compensata da ittero o subittero splenomegalia litiasi biliare

33
H S : dati strumentali Sferociti nello striscio di sangue periferico colorato con May-Grunwald/Giemsa. MCHC >36 Reticolocitosi Test di resistenza osmotica globulare che mostra la completa emolisi delle emazie a concentrazioni di % di NaCl. Analisi delle proteine della membrana eritrocitaria con elettroforesi in gel di poliacrylamide in presenza di sodio dodecyl solfato (SDS-PAGE)
")
34
Sferocitosi Ereditaria
CLINICA Penetranza ed espressività variabili; si presenta spesso nell’infanzia o nell’adolescenzza; esistono classi di gravità diverse (forme gravi, intermedie, silenti). ANEMIA EMOLITICA CRONICA ittero(+/-grave) splenomegalia calcolosi biliare possono comparire ulcere malleolari possono essere presenti alterazioni scheletriche (turricefalia, polidattilia, isodontiasi) CRISI DI DEGLOBULIZZAZIONE emolitiche o crisi aplastiche(deficit di folati, infezioni virali es. parvovirus B19)
. ANEMIA EMOLITICA CRONICA. ittero(+/-grave) splenomegalia. calcolosi biliare. possono comparire ulcere malleolari. possono essere presenti alterazioni scheletriche. (turricefalia, polidattilia, isodontiasi) CRISI DI DEGLOBULIZZAZIONE. emolitiche o crisi aplastiche(deficit di folati, infezioni virali es. parvovirus B19)")
35
Sferocitosi Ereditaria
SVP Hb con MCV normale (sferocitosi) Reticolocitosi (eccetto nelle crisi di deglobulizzazione) Striscio: globuli rossi tutti colorati senza alone chiaro centrale (sferociti) Resistenze globulari ridotte (positività del test dell’autoemolisi corretto da aggiunta di glucosio o test di Simmel) MO Iperplasia eritroblastica
 Reticolocitosi (eccetto nelle crisi di deglobulizzazione) Striscio: globuli rossi tutti colorati senza alone chiaro centrale (sferociti) Resistenze globulari ridotte. (positività del test dell’autoemolisi corretto da aggiunta di glucosio o test di Simmel) MO. Iperplasia eritroblastica.")
36
Sferocitosi Ereditaria
DIAGNOSI Familiarità Caratteristiche emolitiche dell’anemia Negatività del test di Coombs Tipica morfologia allo striscio SVP Test di fragilità osmotica +/- studio delle proteine di membrana (E.F.) PROGNOSI Più severa nella prima infanzia (forme gravi)
 PROGNOSI. Più severa nella prima infanzia (forme gravi)")
37
H S : terapia La splenectomia è la terapia elettiva.
La splenectomia è indicata nei casi di HS caratterizzati da anemia di grado elevato, da emolisi significativa (reticolocitosi > 5%) con conseguente ipersplenismo ed espansione del tessuto midollare a livello anche delle ossa piatte. In età pediatrica va eseguita tra i 5- 9 di età. La splenectomia va presa in considerazione nei soggetti adulti che si sottopongono a colecistectomia per calcolosi biliare. La splenectomia deve essere preceduta da vaccinazione per meningococco, pneumococco, hemophilus influenza.
 con conseguente ipersplenismo ed espansione del tessuto midollare a livello anche delle ossa piatte. In età pediatrica va eseguita tra i 5- 9 di età. La splenectomia va presa in considerazione nei soggetti adulti che si sottopongono a colecistectomia per calcolosi biliare. La splenectomia deve essere preceduta da vaccinazione per meningococco, pneumococco, hemophilus influenza.")
38
ELLISSOCITOSI

39
Ellissocitosi Ereditaria ( HE)
Anemia emolitica autosomica dominante a penetranza completa dovuta ad un difetto della α-spectrina, incapace di associarsi per formare le strutture tetrameriche, per cui le emazie sono incapaci di mantenere forma biconcava dopo diversi passaggi nei capillari. Diffusa nei soggetti di origine africana Il quadro clinico è eterogeneo: nell’ambito di una stessa famiglia si osservano soggetti con forme silenti ben compensate e soggetti con forme moderate o gravi di anemie emolitiche.

40
Ellissocitosi Ereditaria ( HE)
Diagnosi : presenza nello striscio di sangue periferico di emazie a forma ellittica (ellissociti) o ovalare superiore al 10%. Reticolocitosi Splenomegalia Terapia : Splenectomia con le stesse indicazioni della HS
 o ovalare superiore al 10%. Reticolocitosi. Splenomegalia. Terapia : Splenectomia con le stesse indicazioni della HS.")
41
ANEMIE EMOLITICHE DA DIFETTO ENZIMATICO

42
Anemie Emolitiche Enzimopeniche
L’energia necessaria per un eritrocita maturo è fornita solamente dal METABOLISMO DEL GLUCOSIO attraverso due vie La via anaerobica (Embden Meyerhof) La via dei pentosi (shunt dell’esosomonofosfato) Il fabbisogno energetico è necessario per La pompa ionica (ATP) Il sistema riduttivo della metaemoglobina, che mantiene il Fe bivalente (NADH) Il sistema della glutatione reduttasi necessario per proteggre i gruppi tiolici dell’Hb La produzione di 2,3-difosfoglicerato che funzione da modulatore dell’affinità dell’Hb per l’O2 La sintesi del NAD da acido nicotinico, glutamina,glucosio e fosfati inorganici. Con l’invecchiamento degli eritrociti si ha una riduzione dell’attività dei vari enzimi, con aumento del Na, riduzione del K, aumento della meta-Hb, aumentata suscettibilità agli agenti ossidanti, emolisi
 La via dei pentosi (shunt dell’esosomonofosfato) Il fabbisogno energetico è necessario per. La pompa ionica (ATP) Il sistema riduttivo della metaemoglobina, che mantiene il Fe bivalente (NADH) Il sistema della glutatione reduttasi necessario per proteggre i gruppi tiolici dell’Hb. La produzione di 2,3-difosfoglicerato che funzione da modulatore dell’affinità dell’Hb per l’O2. La sintesi del NAD da acido nicotinico, glutamina,glucosio e fosfati inorganici. Con l’invecchiamento degli eritrociti si ha una riduzione dell’attività dei vari enzimi, con. aumento del Na, riduzione del K, aumento della meta-Hb, aumentata suscettibilità agli agenti ossidanti, emolisi.")
43
Anemia emolitica da difetto enzimatico
Anemie congenite dovute a difetto di enzimi eritrocitari, implicati nelle principali vie metaboliche deputate alla produzione di energia per il mantenimento della stabilità dell’emoglobina e delle proteine di membrana. Enzimi dello shunt dei pentoso-fosfati : glucoso-6-fosfato-deidrogenasi (G6PD). Enzimi della glicolisi anaerobia : piruvato-kinasi (PK)
. Enzimi della glicolisi anaerobia : piruvato-kinasi (PK)")
44
Difetti Congeniti Del Metabolismo Energetico
Sono conosciute molte mutanti del metabolismo dei globuli rossi: il 95% è costituito dalle CARENZE DELLA G-6-PD Clinicamente la carenza di G-6-PD è la più frequente FARMACI sulfamidici, cloramfenicolo, primachina, chinina, chinidina acido acetilsalicilico, antipirina, fenacetina, acetanilide, vitamina K AGENTI VARI fave, polline di fave, piselli INFEZIONI virali e batteriche UREMIA, CHETOACIDOSI DIABETICA

45
clinicamente e geneticamente ETEROGENEA
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo Condizione EMOLITICA EREDITARIA clinicamente e geneticamente ETEROGENEA determinata da ridotta capacità delle emazie di resistere al danno ossidativo con accorciamento della vita media

46
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo
Sono conosciute più di 300 mutanti ( G-6-PD-A, G-6-PD-B) Modalità di trasmissione DIAGINICA (per lo più maschi malati) Rara la vera carenza ( delezioni); più frequenti le VARIANTI CON ALTERATTA MOBILITA’ ELETTROFORETICA (mutazioni puntiformi)
 Modalità di trasmissione DIAGINICA (per lo più maschi malati) Rara la vera carenza ( delezioni); più frequenti le VARIANTI. CON ALTERATTA MOBILITA’ ELETTROFORETICA. (mutazioni puntiformi)")
47
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo
FISIOPATOLOGIA (1) Gli eritrociti presentano un deficit di G-6-PD (deficit enzimatico intraglobulare), ma vanno incontro a EMOLISI SOLO SE esposti allo STRESS OSSIDATIVO di alcuni farmaci (sulfamidici, primachina…) o dei componenti delle fave.
 Gli eritrociti presentano. un deficit di G-6-PD (deficit enzimatico intraglobulare), ma vanno incontro a EMOLISI SOLO SE. esposti allo STRESS OSSIDATIVO di alcuni farmaci (sulfamidici, primachina…) o dei componenti delle fave.")
48
dose o concentrazione della noxa ossidante
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo FISIOPATOLOGIA (2) L’aumentata ossidazione dell’Hb dipende da :invecchiamento dei globuli rossi (G-6-PD) variante genetica della G-6-PD (variante mediterraneaforma gravefavismo) dose o concentrazione della noxa ossidante Come conseguenza dell’ossidazione l’Hb tende a precipitare (corpi di Heinz) emolisi per lo più SPLENICA
 L’aumentata ossidazione dell’Hb dipende da :invecchiamento dei globuli rossi (G-6-PD) variante genetica della G-6-PD (variante mediterraneaforma gravefavismo) dose o concentrazione della noxa ossidante. Come conseguenza dell’ossidazione l’Hb tende a precipitare (corpi di Heinz) emolisi per lo più SPLENICA.")
49
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo
CLINICA Forma congenita a esordio precoce: ittero neonatale grave simulante la MEN Forma congenita a esordio giovanile: emolisi cronica con acutizzazioni in caso di assunzione di agenti ossidanti.

50
Trattasi generalmente di crisi emolitiche acute , caratterizzate da
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo CLINICA Trattasi generalmente di crisi emolitiche acute , caratterizzate da Pallore, ittero ingravescente, febbre, dolori lombari, vomito, dolori addominali intensi , più raramente insufficienza renale, che si presentano dopo l’ingestione di farmaci o fave.

51
Anemia severa normocitica, normocromica
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo LABORATORIO Anemia severa normocitica, normocromica Reticolocitosi elevata, della bilirubina indiretta Striscio SVP: numerosi corpi di Heinz intraeritrocitari Carenza della G-6-PD eritrocitaria (determinazione spettrofotometrica)
")
52
Anamnesi (N.B. familiarità, trasmissione diaginica)
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo DIAGNOSI Anamnesi (N.B. familiarità, trasmissione diaginica) Anemia CRONICA con crisi emolitiche scatenate da farmaci/fave; a volte non identificabile l’agente scatenante Reticolocitosi,aumento della bilirubina indiretta-Corpi di Heinz intraeritrocitari Livelli di G-6-PD ridotti (dosaggio enzima) DIAGNOSI DIFFERENZIALE Emolisi acuta a diversa patogenesi
 Anemia CRONICA con crisi emolitiche scatenate da farmaci/fave; a volte non identificabile l’agente scatenante. Reticolocitosi,aumento della bilirubina indiretta-Corpi di Heinz intraeritrocitari. Livelli di G-6-PD ridotti (dosaggio enzima) DIAGNOSI DIFFERENZIALE. Emolisi acuta a diversa patogenesi.")
53
Anamnesi (N.B. familiarità, trasmissione diaginica)
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo DIAGNOSI Anamnesi (N.B. familiarità, trasmissione diaginica) Anemia CRONICA con crisi emolitiche scatenate da farmaci/fave; a volte non identificabile l’agente scatenante Reticolocitosi,aumento della bilirubina indiretta-Corpi di Heinz intraeritrocitari Livelli di G-6-PD ridotti (dosaggio enzima) DIAGNOSI DIFFERENZIALE Emolisi acuta a diversa patogenesi
 Anemia CRONICA con crisi emolitiche scatenate da farmaci/fave; a volte non identificabile l’agente scatenante. Reticolocitosi,aumento della bilirubina indiretta-Corpi di Heinz intraeritrocitari. Livelli di G-6-PD ridotti (dosaggio enzima) DIAGNOSI DIFFERENZIALE. Emolisi acuta a diversa patogenesi.")
54
Carenza di Glucosio-6-Fosfato Deidrogenasi Anemia primachina sensibile e Favismo
DECORSO E PROGNOSI 5-10% dei pazienti in caso di crisi emolitica da fave va incontro a insufficienza renale acuta. Meno grave il quadro dopo l’ingestione di farmaci. TERAPIA Profilattica. Evitare l’assunzione di farmaci ossidanti e di fave Supporto durante le crisi emolitiche: promuovere abbondante diuresi Trasfusioni

55
Agenti che possono causare emolisi nel deficit di G-6-PD
Fave, piselli Farmaci Sulfamidici sulfoni (cotrimoxazolo, salazopirina, sulfacetamide, sulfanilamide,sulfapiridina,…) Antibatterici (cloramfenicolo, nitrofurantoina, acido peraminosalicilico…) Antimalarici (primachina, clorochina) Analgesici (acido acetilsalicilico, acetofenacetina, piramidone..) Altri ( vit.K, blu di metilene, probenecid, acido nalidixico, niridazolo…) Infezioni virali e batteriche, chetoacidosi
 Antibatterici (cloramfenicolo, nitrofurantoina, acido peraminosalicilico…) Antimalarici (primachina, clorochina) Analgesici (acido acetilsalicilico, acetofenacetina, piramidone..) Altri ( vit.K, blu di metilene, probenecid, acido nalidixico, niridazolo…) Infezioni virali e batteriche, chetoacidosi.")
56
Anemia da deficit G6PD Anemia emolitica congenita X-linked, per cui sono colpiti i maschi. E’ rara la condizione di omozigosi femminile, mentre le donne eterozigoti avranno una doppia popolazione eritrocitaria. La mutazione del gene codificante per la G6PD sembra sia un vantaggio selettivo: la crescita dei parassiti malarici è inibita nelle emazie prive dell’enzima. Tale mutazione si associa nelle popolazioni negre alla mutazione puntiforme (6) del gene globinico del cromosoma 11 per l’effetto positivo sullo sviluppo della malattia falcemica. La riduzione dell’attività enzimatica varia dal 5% al 50% Il deficit di G6PD causa una ridotta formazione di NADPH con conseguente riduzione delle reduttasi e superossido-dismutasi NADPH dipendenti. L’emivita eritrocitaria si riduce per il prevalere dei processi ossidativi sia a livello della membrana sia a livello dell’emoglobina, soprattutto in presenza di agenti chimici ossidanti. L’emoglobina ossidata forma i corpi di Heinz; le emazie con i corpi di Heinz sono sequestrate a livello splenico.
 del gene globinico del cromosoma 11 per l’effetto positivo sullo sviluppo della malattia falcemica. La riduzione dell’attività enzimatica varia dal 5% al 50% Il deficit di G6PD causa una ridotta formazione di NADPH con conseguente riduzione delle reduttasi e superossido-dismutasi NADPH dipendenti. L’emivita eritrocitaria si riduce per il prevalere dei processi ossidativi sia a livello della membrana sia a livello dell’emoglobina, soprattutto in presenza di agenti chimici ossidanti. L’emoglobina ossidata forma i corpi di Heinz; le emazie con i corpi di Heinz sono sequestrate a livello splenico.")
57
Cause di crisi emolitiche in presenza di deficit di G6PD
Infezioni Ingestione di fave o piselli Farmaci : blu di metilene, salicilati, cloramfenicolo, primachina, acido nalidixico, sulfametoxazolo, sulfanilamide, isosorbide dinitrato, fenacetina, doxorubicina. Sostanze chimiche: naftalina, nitrati e nitriti, fenilidralazina, trinitrotoluene.

58
Diagnosi di deficit di G6PD
Familiarità Dosaggio attività enzimatica della G6PD mediante test di riduzione del NADP+ a NAPDH (<30%). Test di riduzione della metemoglobina dopo esposizione dell’eritrocita al blu di metilene.
. Test di riduzione della metemoglobina dopo esposizione dell’eritrocita al blu di metilene.")
59
Forme cliniche da deficit di G6PD
Forma congenita a esordio precoce che mima malattia emolitica neonatale (MEN) Forma congenita a esordio giovanile : la crisi emolitica insorge in seguito ad assunzione di sostanze ossidanti. Favismo : crisi emolitica acuta in seguito ad assunzione, raramente ad esposizione, di fave
 Forma congenita a esordio giovanile : la crisi emolitica insorge in seguito ad assunzione di sostanze ossidanti. Favismo : crisi emolitica acuta in seguito ad assunzione, raramente ad esposizione, di fave.")
60
Anemia emolitica da deficit di Piruvato Kinasi (PK)
Anemia congenita autosomica recessiva con manifestazioni cliniche solo nei soggetti omozigoti. La mutazione del gene PK-LR, situato sul cromosoma 1, causa il difetto dell’enzima piruvato kinasi essenziale nella glicolisi anaerobica per la sintesi di ATP. Il conseguente deficit energetico è responsabile della ridotta attività della pompa Na/K e della conservazione dell’integrità della membrana eritrocitaria. Il blocco della via glicolitica anaerobica causa un accumulo di 2,3-difosfoglicerato (2,3-DPG) con conseguente spostamento a destra della curva di dissociazione dell’emoglobina I soggetti omozigoti presentano anemia subittero e splenomegalia.
 con conseguente spostamento a destra della curva di dissociazione dell’emoglobina. I soggetti omozigoti presentano anemia subittero e splenomegalia.")
61
Carenza di Piruvato Kinasi (PK) e di altri Enzimi
Condizione EMOLITICA CRONICA EREDITARIA , da ridotta disponibilità di ATP, con conseguente ridotta sopravvivenza eritrocitaria Forme rare La PK costituisce il 95% delle forme non da di G-6-PD Trasmissione AUTOSOMICA RECESSIVA La PK è necessaria per produrre ATP, che è necessario a sua volta per la pompa del sodio I globuli rossi giovani ed i reticolociti soffrono meno la carenza Gli OMOZIGOTI manifestano anemia e ittero dall’ infanzia , quadro clinico e di labotatorio tipico dell’emolisi cronica. I farmaci non sono implicati. Possibili crisi di deglobulizzazione

62
Carenza di Piruvato Kinasi (PK)
DIAGNOSI Anemia cronica Reticolocitosi, iperbilirubinemia indiretta Splenomegalia Dosaggio PK

63
Carenza di Piruvato Kinasi (PK) e di altri Enzimi
TERAPIA 1) Splenectomia dopo i 5-10 anni, in caso di anemia sintomatica; si assiste a: marcato incremento dei reticolociti ( fino ad oltre il 50%) e ad un miglioramento del quadro clinico e laboratoristico. 2) Folati per os. 3) Trasfusioni di emazie concentrate nei casi con anemia grave (Hb < 8 g/dl).
 Splenectomia dopo i 5-10 anni, in caso di anemia sintomatica; si assiste a: marcato incremento dei reticolociti ( fino ad oltre il 50%) e ad un miglioramento del quadro clinico e laboratoristico. 2) Folati per os. 3) Trasfusioni di emazie concentrate nei casi con anemia grave (Hb < 8 g/dl).")
64
Carenza di Piruvato Kinasi (PK) e di altri Enzimi
Condizione EMOLITICA CRONICA EREDITARIA , da ridotta disponibilità di ATP, con conseguente ridotta sopravvivenza eritrocitaria Forme rare La PK costituisce il 95% delle forme non da di G-6-PD Trasmissione AUTOSOMICA RECESSIVA La PK è necessaria per produrre ATP, che è necessario a sua volta per la pompa del sodio I globuli rossi giovani ed i reticolociti soffrono meno la carenza Gli OMOZIGOTI manifestano anemia e ittero dall’ infanzia , quadro clinico e di labotatorio tipico dell’emolisi cronica. I farmaci non sono implicati. Possibili crisi di deglobulizzazione Striscio SVP: Emazie crenate, modesta anisopoichilocitosi, acantociti Diagnosi: dosaggio dell’attività enzimatica Terapia: supporto trasfusionale associato a terapia chelante ove necessario Splenectomia nei casi con emolisi grave

65
Terapia anemie emolitiche congenite
Prevenzione delle crisi conl’astensione di farmaci ossidanti, fave e piselli. Terapia trasfusionale e/o ferro-chelante nei casi di crisi emolitiche di notevole entità associata ad iperidratazione. Splenectomia nei casi di gravi e ripetute crisi emolitiche

66
ANEMIA EMOLITICA AUTOIMMUNE

67
Anemia emolitica autoimmune (AEA) Emolisi da anticorpi anti-eritrociti
AEA è una malattia con incidenza di 10 casi per milione ed è più frequente nelle donne che negli uomini Le AEA si possono distinguere per: - eziologia: primitiva/secondaria - età del paziente - carattere anticorpi: (autoanticorpi / isoanticorpi ) - isotipo anticorpi: (Ig G / Ig M) -temperatura attività anticorpale: (caldi, freddi e bifasici) -sede dell’emolisi: (intravascolare / extravascolare)
 - isotipo anticorpi: (Ig G / Ig M) -temperatura attività anticorpale: (caldi, freddi e bifasici) -sede dell’emolisi: (intravascolare / extravascolare)")
68
AEA: eziologia Fattori fisici: ustioni, traumi da corsa o marcia, protesi valvolari. Fattori chimici: prodotti arsenicali e metilici Agenti infettivi: diplococco pneumoniae, mycoplasma, CMV, EBV,HIV, ecc. Farmaci: metildopa-levodopa Patologie: linfoproliferative, neoplastiche, connettivali Incompatibilità ABO Incompatibilità Rh

69
A E A : età Bambini : forme secondarie ad insorgenza acuta, spesso con risoluzione completa Adulti : forme primitive, spesso con andamento cronico per risposte terapeutiche parziali.

70
Anemie Emolitiche Autoimmuni
CLASSIFICAZIONE In rapporto alla presenza o assenza di una malattia di base : Primitive (idiopatiche) (50%) Secondarie malattie linfoproliferative croniche neoplasie solide infezioni connettiviti malattie infiammatorie croniche epatopatie croniche farmaci ( alfa metil dopa)
 (50%) Secondarie -malattie linfoproliferative croniche -neoplasie solide -infezioni -connettiviti -malattie infiammatorie croniche -epatopatie croniche -farmaci ( alfa metil dopa)")
71
Anemie Emolitiche Autoimmuni
Anemia dovuta a distruzione eritrocitaria da autoanticorpi CLASSIFICAZIONE In rapporto alle caratteristiche sierologiche AEA da anticorpi caldi (80-90%) AEA da anticorpi freddi (10-20%)
 AEA da anticorpi freddi (10-20%)")
72
Anemie Emolitiche Autoimmuni
Anemia dovuta a distruzione eritrocitaria da autoanticorpi CLASSIFICAZIONE In rapporto alle caratteristiche sierologiche AEA da anticorpi caldi (80-90%) AEA da anticorpi freddi (10-20%)
 AEA da anticorpi freddi (10-20%)")
73
Immunologia AEA Gli anticorpi anti-eritrocitari possono essere:
IgG vs antigeni gruppo specifici (Rh, P, M, N…) IgM vs antigeni comuni ( I, i ) Anticorpi caldi ( >IgG) º C Anticorpi freddi (>IgM) ° C Anticorpi Bi-fasici (emolisina di Donald Landsteiner) IgG anti P º C fissazione ° C emolisi Anticorpi completi (>IgM) agglutinazione con emazie in soluzione fisiologica Anticorpi incompleti (>IgG) agglutinazione con emazie in mezzo albuminoideo Agglutinine agglutinazione in vitro senza emolisi in vivo Emolisine emolisi in vitro o in vivo per attivazione del complemento
 IgM vs antigeni comuni ( I, i ) Anticorpi caldi ( >IgG) º C. Anticorpi freddi (>IgM) 0-27 ° C. Anticorpi Bi-fasici (emolisina di Donald Landsteiner) IgG anti P 4-27 º C fissazione 34-37° C emolisi. Anticorpi completi (>IgM) agglutinazione con emazie in soluzione fisiologica. Anticorpi incompleti (>IgG) agglutinazione con emazie in mezzo albuminoideo. Agglutinine agglutinazione in vitro senza emolisi in vivo. Emolisine emolisi in vitro o in vivo per attivazione del complemento.")
74
Eziologia e Patogenesi
Gli anticorpi sono rivolti spesso contro un antigene identificabile Esiste un rapporto inverso tra quantità di anticorpi sulle emazie e sopravvivenza eritrocitaria AEA da anticorpi caldi Si ha ridotta sopravvivenza eritrocitaria in rapporto all’azione macrofagica( cordoni di Billroth e cellule di Kuppfer) Azione del sistema macrofagico: sferocitosi e successiva frammentazione delle emazie; sulla membrana eritrocitaria, con le IgG si possono avere frazioni del complemento (C3) favorenti l’ opsonizzazione Prima si ha interazione fra macrofagi ed emazie sensibilizzate dal legame fra Fc di IgG e suo recettore. Segue fagocitosi con perdita di estese parti della membrana eritrocitaria il grado di sferocitosi correla con l’entità dell’emolisi Rara è l’emolisi intra-vascolare
 Azione del sistema macrofagico: sferocitosi e successiva frammentazione delle emazie; sulla membrana eritrocitaria, con le IgG si possono avere frazioni del complemento (C3) favorenti l’ opsonizzazione. Prima si ha interazione fra macrofagi ed emazie sensibilizzate dal legame fra Fc di IgG e suo recettore. Segue fagocitosi con perdita di estese parti della membrana eritrocitaria. il grado di sferocitosi correla con l’entità dell’emolisi. Rara è l’emolisi intra-vascolare.")
75
Eziologia e Patogenesi
AEA da anticorpi freddi La distruzione eritrocitaria è dovuta ad anticorpi di tipo IgM L’emolisi è in rapporto alla capacità di attivazione completa della sequenza del complemento (C1-C9) sulla superficie eritrocitaria Ampiezza termica degli anticorpi da 20 a 25° C: emolisi a temperature inferiori: agglutinazione Si ha sensibilizzazione eritrocitaria nel microcircolo con lisi intravascolare o sequestro e lisi nella milza e nel fegato La lisi eritrocitaria è diretta o per opsonizzazione macrofagica Si ha il fenomeno della acrocianosi (aggregazione eritrocitaria nel microcircolo) in rapporto alla temperatura esterna
 sulla superficie eritrocitaria. Ampiezza termica degli anticorpi da 20 a 25° C: emolisi. a temperature inferiori: agglutinazione. Si ha sensibilizzazione eritrocitaria nel microcircolo con lisi intravascolare o sequestro e lisi nella milza e nel fegato. La lisi eritrocitaria è diretta o per opsonizzazione macrofagica. Si ha il fenomeno della acrocianosi (aggregazione eritrocitaria nel microcircolo) in rapporto alla temperatura esterna.")
76
Eziologia e Patogenesi
Fino agli anni ’30 difficoltà a differenziare le anemie emolitiche congenite da quelle acquisite L’introduzione del test di COOMBS (1945) basato sull’uso del siero di coniglio anti-IgG umane e/o frazioni del C , ha reso possibile la suddetta distinzione. Lo studio della SOPRAVVIVENZA ERITROCITARIA ha introdotto il concetto di: -anemia emolitica da difetto intracorpuscolare i GR hanno una sopravvivenza ridotta sia nei pazienti con AEA che nei soggetti normali, una volta trasfusi. -anemia emolitica da difetto extracorpuscolare i GR sopravvivono più a lungo se infusi nei soggetti normali
 basato sull’uso del siero di coniglio anti-IgG umane e/o frazioni del C , ha reso possibile la suddetta distinzione. Lo studio della SOPRAVVIVENZA ERITROCITARIA ha introdotto il concetto di: -anemia emolitica da difetto intracorpuscolare. i GR hanno una sopravvivenza ridotta sia nei pazienti con AEA che nei soggetti normali, una volta trasfusi. -anemia emolitica da difetto extracorpuscolare. i GR sopravvivono più a lungo se infusi nei soggetti normali.")
77
Emolisi intravascolare
L’emolisi avviene in presenza di elevati titoli di anticorpi freddi o bi - fasici, che attivano il complemento. Le frazioni C5-C9 del complemento inducono soluzione di continuo della membrana degli eritrociti con ingresso di acqua. La perfrigerazione è essenziale per il legame anticorpo-emazia. L’emoglobina liberata si fissa all’aptoglobina. Emoglobinuria insorge se l’aptoglobina disponibile è totalmente saturata dall’ Hb liberata dagli eritrociti lisati.

78
Emolisi extravascolare
L’ emolisi eritrocitaria è sostenuta da anticorpi completi agglutinanti o da anticorpi incompleti incapaci di attivare il complemento. In presenza di anticorpi agglutinanti, il sequestro del complesso eritrocita- anticorpo è a livello prevalentemente epatico. In presenza di anticorpi incompleti, le emazie legate con le IgG1 sono sequestrate a livello splenico, dove i macrofagi posseggono i recettori per il frammento Fc delle IgG.

79
Test di Coombs Diretto: positivo per presenza di anticorpi (Ab) completi, IgM, capaci di agglutinare. Indiretto: positivo per presenza di anticorpi incompleti IgG.

80
Caratteristiche anticorpi caldi
Autoanticorpi (Ig G) Sottoclasse Ig G1 Optimum termico 37°C ( range 0°- 40°) Diretti antigene “e” del sistema Rh Incapaci di attivare il complemento Incompleti perché non agglutinano emazie Emolisi extravascolare
 Sottoclasse Ig G1. Optimum termico 37°C ( range 0°- 40°) Diretti antigene e del sistema Rh. Incapaci di attivare il complemento. Incompleti perché non agglutinano emazie. Emolisi extravascolare.")
81
AEA da anticorpi caldi Esordio
CLINICA Esordio LENTO ed insidioso con prevalenza dei sintomi dell’anemia ACUTO, con pallore marcato, subittero, febbre, epatosplenomegalia. Nelle forme gravi :polipnea, tachicardia Forme SECONDARIE: spesso prevalenti i sintomi della malattia di base (malattie linfoproliferative, connettiviti); possibili epatosplenomegalia e/o adenomegalie marcate IDIOPATICHE esame obiettivo negativo a parte i segni dell’anemia, epato-splenomegalia possibile nelle forme gravi.
; possibili epatosplenomegalia e/o adenomegalie marcate. IDIOPATICHE esame obiettivo negativo a parte i segni dell’anemia, epato-splenomegalia possibile nelle forme gravi.")
82
AEA da anticorpi caldi LABORATORIO - anemia di grado variabile
- sferocitosi, macrocitosi, policromasia - reticolocitosi, eritroblasti circolanti - LDH e bilirubina indiretta - M.O.: (iperplasia eritroblastica +/- marcata con prevalenza delle forme mature)
")
83
AEA da anticorpi caldi DIAGNOSI TEST DI COOMBS DIRETTO
Dimostrazione di Ig e/o C sulle emazie con sieri anti-globuline sempre più selettivi (globale,gamma e non-gamma, monospecifici per le classi di IgG ed il C3 e C4) E’ possibile riscontrare -solo IgG -IgG+C3/C4 -solo C3/C4 -IgA o IgM+IgG+C3/C4
 E’ possibile riscontrare. -solo IgG. -IgG+C3/C4. -solo C3/C4. -IgA o IgM+IgG+C3/C4.")
84
AEA da anticorpi caldi TEST DI COOMBS INDIRETTO
DIAGNOSI TEST DI COOMBS INDIRETTO Gli anticorpi possono essere dimostrati anche o solo nel siero -Il test di Coombs diretto è essenziale -Il test di Coombs indiretto(presenza di Ab solo nel siero) può indicare sensibilizzazione passiva (trasfusionale o materna) -E’ possibile determinare quantitativamente le moli di Ig adese alle emazie ( utile per le forme più lievi)
 può indicare sensibilizzazione passiva (trasfusionale o materna) -E’ possibile determinare quantitativamente le moli di Ig adese alle emazie ( utile per le forme più lievi)")
85
AEA da anticorpi caldi SPLENECTOMIA IMMUNOSOPPRESSORI TERAPIA (2)
Nei pz recidivati dopo terapia CCS o durante dosi di mantenimento elevate. Remissione completa o parziale 2/3 dei casi. IMMUNOSOPPRESSORI Nei pz non rispondenti ai CCS e/o alla splenectomia (azatioprina)
")
86
AEA da anticorpi caldi TERAPIA (1) CORTICOSTEROIDI
La terapia più efficace ( riducono o aboliscono l’emolisi nei 2/3 dei pazienti) Efficacia dose-dipendente, maggiore nelle connettiviti. (10% dei pz non risponde) Dosaggio elevato PDN 1-3 mg/Kg/die per giorni, poi ridurre e proseguire per 3-4 mesi. La recidiva è frequente TRASFUSIONI Limitate ai pz con rischio cardiologico elevato e/o in pericolo di vita per la gravissima anemia (Hb< 4 g/dl) (difficoltà di ottenere prove crociate libere, ridotta emivita delle emazie trasfuse). E’ raro trovare donatori compatibili; la trasfusione, se necessaria, può essere eseguita con emazie parzialmente compatibili ed effettuata molto lentamente.
 Efficacia dose-dipendente, maggiore nelle connettiviti. (10% dei pz non risponde) Dosaggio elevato PDN 1-3 mg/Kg/die per giorni, poi ridurre e proseguire per 3-4 mesi. La recidiva è frequente. TRASFUSIONI. Limitate ai pz con rischio cardiologico elevato e/o in pericolo di vita per la gravissima anemia (Hb< 4 g/dl) (difficoltà di ottenere prove crociate libere, ridotta emivita delle emazie trasfuse). E’ raro trovare donatori compatibili; la trasfusione, se necessaria, può essere eseguita con emazie parzialmente compatibili ed effettuata molto lentamente.")
87
AEA da anticorpi caldi DECORSO E SOPRAVVIVENZA
-Decorso variabile (lunghe remissioni o frequenti recidive) -Mortalità bassa con sopravvivenza a 10 anni >75% -Nelle forme secondarie la prognosi e la sopravvivenza dipendono dalla malattia di base -Nei bambini la prognosi è migliore (AEA spesso correlata ad infezioni virali)
 -Mortalità bassa con sopravvivenza a 10 anni >75% -Nelle forme secondarie la prognosi e la sopravvivenza dipendono dalla malattia di base. -Nei bambini la prognosi è migliore (AEA spesso correlata ad infezioni virali)")
88
Caratteristiche anticorpi freddi
Ig M in genere monoclonali ristretti per catene K Diretti contro antigene “I” Completi con attività agglutinante Optimum termico 0° - 4° C Fissano eritrociti a temperature >15° e <32° C Attivano complemento Fissano frazione C3 e C4 del complemento Emolisi intravasale

89
AEA da anticorpi freddi
Malattie emolitiche autoimmuni da anticorpi che agiscono a temperatura < 37º C (usualmente inf. a 31º C) Forme rare, sintomatologia acuta o più raramente cronica Lesione eritrocitaria mediata da attivazione del C sulla superficie delle emazie -1905: agglutinine fredde descritte da Donald e Landstainer -1918: descritta la sindrome emolitica in corso di polmonite -1925: descritta l’associazione fra crioagglutinine e sindrome acrocianotica, tipo Raynaud
 Forme rare, sintomatologia acuta o più raramente cronica. Lesione eritrocitaria mediata da attivazione del C sulla superficie delle emazie : agglutinine fredde descritte da Donald e Landstainer : descritta la sindrome emolitica in corso di polmonite : descritta l’associazione fra crioagglutinine e sindrome acrocianotica, tipo Raynaud.")
90
AEA da anticorpi freddi
EZIOLOGIA IDIOPATICHE Picco di incidenza età >60 anni; in alcuni casi si tratta di forme simili alla macroglobulinemia di Waldenstrom (crioagglutininemie) SECONDARIE Le più frequenti sono caratterizzate da processo emolitico acuto, self- limited, in corso di: -polmonite atipica da Mycoplasma pneumoniae (PAP) -mononucleosi in adolescenti o giovani adulti (virus Epstein-Barr).
 SECONDARIE. Le più frequenti sono caratterizzate da processo emolitico acuto, self- limited, in corso di: -polmonite atipica da Mycoplasma pneumoniae (PAP) -mononucleosi in adolescenti o giovani adulti (virus Epstein-Barr).")
91
AEA da anticorpi freddi
PATOGENESI Sviluppo di crioagglutinine con specificità o reattività crociata per antigeni eritrocitari (es.I/i) PRIMARIO (monoclonale) SECONDARIO Disturbi anticorpopoiesi(monoclonale) (es. Malattie linfoprolifertive) Microrganismi (policlonale) (es. Mycoplasma, EBV) Le crioagglutinine (>IgM) legano ed attivano il C a basse temperature
 PRIMARIO (monoclonale) SECONDARIO Disturbi anticorpopoiesi(monoclonale) (es. Malattie linfoprolifertive) Microrganismi (policlonale) (es. Mycoplasma, EBV) Le crioagglutinine (>IgM) legano ed attivano il C a basse temperature.")
92
AEA da anticorpi freddi
INCIDENZA, CLNICA E LABORATORIO Sono forme rare(10-20% delle AEA), più frequenti nel sesso femminile Nella maggioranza dei pz con polmonite atipica primaria (PAP) e mononucleosi (60%) si possono riscontrare anticorpi freddi ; tuttavia la comparsa di un processo emolitico è rara (<5%). Si possono distinguere: -forme ad andamento cronico +/- ittero -forme acute causate da perfrigerazione -forme recidivanti
, più frequenti nel sesso femminile. Nella maggioranza dei pz con polmonite atipica primaria (PAP) e mononucleosi (60%) si possono riscontrare anticorpi freddi ; tuttavia la comparsa di un processo emolitico è rara (<5%). Si possono distinguere: -forme ad andamento cronico +/- ittero. -forme acute causate da perfrigerazione. -forme recidivanti.")
93
AEA da anticorpi freddi
CARATTERISTICI: -forme da M.pneumoniae (PAP) emolisi acuta in fase di guarigione da polmonite -forme da EBV(mononucleosi) Durante l’infezione (esordio e successivamente) , entro la 3° settimana -forme secondarie a patologie autoimmuni o linfoproliferative Sintomi della malattia primaria -fenomeni di acrocianosi Dita mani e piedi, naso e orecchie -rare manifestazioni ulcerative e necrotiche
 emolisi acuta in fase di guarigione da polmonite. -forme da EBV(mononucleosi) Durante l’infezione (esordio e successivamente) , entro la 3° settimana. -forme secondarie a patologie autoimmuni o linfoproliferative. Sintomi della malattia primaria. -fenomeni di acrocianosi. Dita mani e piedi, naso e orecchie. -rare manifestazioni ulcerative e necrotiche.")
94
AEA da anticorpi freddi
LABORATORIO Anemia: raramente grave (Hb>5-6g/dl), sferocitosi, schistocitosi, policromasia, reticolocitosi Agglutinazione GR in provetta o sullo striscio SVP ( a basse temp., 4° C) emocromo a caldo (37° C) Aptoglobina ridotta Emosiderinuria + (se emolisi intravascolare) Crioagglutinine: IgM (rare IgA o IgG) Coombs diretto positivo con sieri anti-C (abs eliminati dai lavaggi) Anticorpi per lo più anti I/i
, sferocitosi, schistocitosi, policromasia, reticolocitosi. Agglutinazione GR in provetta o sullo striscio SVP ( a basse temp., 4° C) emocromo a caldo (37° C) Aptoglobina ridotta. Emosiderinuria + (se emolisi intravascolare) Crioagglutinine: IgM (rare IgA o IgG) Coombs diretto positivo con sieri anti-C (abs eliminati dai lavaggi) Anticorpi per lo più anti I/i.")
95
AEA da anticorpi freddi
TERAPIA Poco efficace Evitare l’esposizione al freddo Trasfusione (emazie lavate) solo se pericolo di vita Nelle crioagglutininemie gravi: plasmaferesi+ciclofosfamide
 solo se pericolo di vita. Nelle crioagglutininemie gravi: plasmaferesi+ciclofosfamide.")
96
Caratteristiche anticorpi bifasici
Anticorpi IgG Diretti antigene “P” Fissano le emazie a 0°- 20°C Attivano complemento a 37°C

97
EMOGLOBINURIA PAROSSISTICA DA FREDDO
Rara forma di AEA da anticorpi freddi con emolisi massiva da esposizione al freddo Incidenza bassa (2-5% di tutti i casi di AEA) In passato la EP da freddo era associata alla sifilide MANIFESTAZIONI CLINICHE Dopo min o ore dall’esposizione al freddo, dolori lombari, crampi alle gambe, all’addome, febbre, emoglobinuria Durata della crisi: alcune ore, possono associarsi fenomeni tipo Raynaud e orticaria, l’ittero compare successivamente LABORATORIO Coombs diretto + per il C solo durante l’attacco L’anticorpo di Donald Landsteiner NON E’ AGGLUTINANTE, si lega ai GR alle basse temperature nel microcircolo, AGISCE ALLE TEMPERATURE più ALTE Ab diretto contro ag sistema P TERAPIA Evitare l’esposizione al freddo CCS e azatioprina
 In passato la EP da freddo era associata alla sifilide. MANIFESTAZIONI CLINICHE. Dopo min o ore dall’esposizione al freddo, dolori lombari, crampi alle gambe, all’addome, febbre, emoglobinuria. Durata della crisi: alcune ore, possono associarsi fenomeni tipo Raynaud e orticaria, l’ittero compare successivamente. LABORATORIO. Coombs diretto + per il C solo durante l’attacco. L’anticorpo di Donald Landsteiner NON E’ AGGLUTINANTE, si lega ai GR alle basse temperature nel microcircolo, AGISCE ALLE TEMPERATURE più ALTE. Ab diretto contro ag sistema P. TERAPIA. Evitare l’esposizione al freddo. CCS e azatioprina.")
98
A E A : terapia Arrestare emolisi Rallentare formazione anticorpi
Ripristinare livelli Hb idonei

99
A E A : terapia . Steroidi: prednisone 1/2 mg/Kg/die per 4 sett.; la dose va scalata nei rispondenti fino alla dose minima, capace di controllare l’emolisi. Nei soggetti non rispondenti agli steroidi è utile: - aziatioprina: 50/100 mg/die - ciclofosfamide: 50/100 mg/die. La splenectomia va riservata ai pazienti con AEA non rispondenti né agli steroidi né agli immunosoppresori.

Presentazioni simili


















>")
