Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
GESTIONE E GOVERNO DEI DISPOSITIVI MEDICI NELLA PRATICA CLINICA E…NON! Comitato Etico 23.02.2016

2
DISPOSITIVI MEDICI: MATERIA complessa???

3
Perché?? SONO TANTI: si calcola che in Europa circolino circa mezzo milione di DM e questo dato è in continua crescita. Innovazione continua….. LA NORMATIVA è molto complessa: …Europea,….Nazionale…..Regionale ed è continuamente aggiornata I CONFRONTI tra i diversi prodotti della stessa tipologia per le stesse indicazioni d’uso…..sono difficili da fare Non esistono ELENCHI chiari come per le specialità medicinali Non esiste una REGISTRAZIONE Ministeriale ma solo l’iscrizione alla banca dati del M.d.S. Gli STUDI disponibili sono pochissimi

4
E’ con il DPR n. 392 del 1998 che viene differenziato il PMC dal Dispositivo Medico e viene per la 1° volta definito un elenco di prodotti chiamati PMC. Questo elenco si differenzia dai DM che rimangono ancora assoggettati al DPR n. 128/86. Inizialmente sono regolamentati dal TULS del 1934 che li definisce tutti Presidi Medico-Chirurgici esiste un unico elenco che non fa distinzione di categoria, uso ecc… non esiste una definizione normativa di PMC e vengono genericamente definiti come “prodotti a rilevanza sanitaria non riconducibile alla categoria dei medicinali” Nel 1986 con il DPR n. 128 per la 1° volta vengono suddivisi in sottogruppi differenti anche se la distinzione è solo nominale e non giuridica PRESIDI MEDICO-CHIRURGICI: Presidi chimici Dispositivi medici Diagnostici in vitro Cenni storici…..

5
Oggi le direttive Europee li ricomprendono in diverse categorie…..

6
Un inciso per Disinfettanti, antisettici, battericidi, germicidi.. Un disinfettante o un antisettico ecc. può essere classificato come: Specialità medicinale per cute lesa o integra (risponde alla normativa sulle specialità medicinali) Presidio Medico-chirurgico per ambienti e superfici (risponde alla normativa sui PMC) Dispositivo medico per disinfezione DM (risponde alla normativa sui DM)
 Presidio Medico-chirurgico per ambienti e superfici (risponde alla normativa sui PMC) Dispositivo medico per disinfezione DM (risponde alla normativa sui DM).")
7
La commercializzazione di un DM è possibile solo previa apposizione del marchio CE (Conformità Europea) cioè dopo avere dimostrato che risulta conforme ai requisiti essenziali stabiliti dalla norma.(Allegati al DL 46/97 e aggiornamenti) Il Marchio CE è il simbolo di conformità a tutti i requisiti della regolamentazione applicabile al dispositivo. Il Marchio CE viene rilasciato dagli Organismi Notificati (ON) Si tratta di organismi autorizzati, con apposita procedura, dalle autorità competenti dei vari Stati dell'Unione Europea e designati ad espletare le procedure di certificazione. Che cosa fanno: valutano il sistema di qualità controllano il mantenimento del sistema di qualità esaminano la progettazione del prodotto certificano il campione rappresentativo del prodotto VERIFICANO CHE IL DM SIA CONFORME AI REQUISITI ESSENZIALI PREVISTI DALLA NORMATIVA ….alcuni punti fermi…
 Si tratta di organismi autorizzati, con apposita procedura, dalle autorità competenti dei vari Stati dell Unione Europea e designati ad espletare le procedure di certificazione. Che cosa fanno: valutano il sistema di qualità controllano il mantenimento del sistema di qualità esaminano la progettazione del prodotto certificano il campione rappresentativo del prodotto VERIFICANO CHE IL DM SIA CONFORME AI REQUISITI ESSENZIALI PREVISTI DALLA NORMATIVA ….alcuni punti fermi….")
8
Classi di rischio I criteri di classificazione sono contenuti nell’allegato IX del DL 46/97 e aggiornamenti I DM sono raggruppati, in funzione della loro complessità e della pericolosità per il paziente in 4 classi: Classe I Classe IIa Classe IIb Classe III A sua volta la classe I è suddivisa in Is (sterile) e Im ( di misura) La classificazione dipende dalla destinazione d’uso indicata dal fabbricante e va attribuita consultando le regole di classificazione riportate nell’allegato IX del D.L. 46/97 e modifiche Più alta è la classe di rischio tanto maggiori saranno le garanzie di sicurezza che il fabbricante dovrà fornire per la produzione ….alcuni punti fermi…

9
La classe di rischio viene stabilita sulla base di diversi parametri contenuti nell'allegato IX della 46/97 e agg.: INVASIVITA’ Un Dispositivo Medico è invasivo quando penetra, interamente o in parte, nel corpo attraverso un orifizio naturale o attraverso la superficie (sonde, aghi, protesi, ecc.) DIPENDENZA DA UNA FONTE DI ENERGIA (attivi o non attivi) E' attivo ogni Dispositivo Medico la cui azione dipenda da una fonte di energia esterna (pacemaker, defibrillatori ecc.) DURATA CONTINUA DI UTILIZZO : E' il tempo di attività di quel Dispositivo Medico: breve < di 60’ (es. ago, siringa, medicazione) media < di 30gg (es. catetere urinario, sonda NG,CVC a breve permanenza, ecc...) lunga > di 30gg (es. port, protesi, stent) ….alcuni punti fermi…
 media < di 30gg (es. catetere urinario, sonda NG,CVC a breve permanenza, ecc...) lunga > di 30gg (es. port, protesi, stent) ….alcuni punti fermi….")
10
CLASSE I es. siringhe senz’ago CLASSE IIa es. siringhe con ago CLASSE IIb es. suture non riassorbibili CLASSE III es. protesi ortopediche, pacemakers

11
I Dispositivi Medici vengono identificati da due parametri: CND - Classificazione Nazionale Dispositivi Numero di Repertorio - numero di iscrizione del DM alla banca dati del Ministero della Salute ….alcuni punti fermi…

12
ATC ( classificazione anatomico, terapeutico e chimico ) J-ANTIMICROBICI GENERALI PER USO SISTEMICO J01-ANTIBATTERICI PER USO SISTEMICO J01C-ANTIBATTERICI BETA-LATTAMICI PENICILLINE J01CA-PENICILLINE AMPIO SPETTRO JO1CA01-AMPICILLINA 020121087-AMPLITAL 1G FIALE JO1CA04-AMOXICILLINA 023086150-ZIMOX 1GR CP
 J-ANTIMICROBICI GENERALI PER USO SISTEMICO J01-ANTIBATTERICI PER USO SISTEMICO J01C-ANTIBATTERICI BETA-LATTAMICI PENICILLINE J01CA-PENICILLINE AMPIO SPETTRO JO1CA01-AMPICILLINA AMPLITAL 1G FIALE JO1CA04-AMOXICILLINA ZIMOX 1GR CP")
13
Categoria CNDDescrizione categoria CND ADISPOSITIVI DA SOMMINISTRAZIONE, PRELIEVO E RACCOLTA BDISPOSITIVI PER EMOTRASFUSIONE ED EMATOLOGIA CDISPOSITIVI PER APPARATO CARDIOCIRCOLATORIO DDISINFETTANTI, ANTISETTICI E PROTEOLITICI (D. Lgs. 46/97) FDISPOSITIVI PER DIALISI GDISPOSITIVI PER APPARATO GASTROINTESTINALE HDISPOSITIVI DA SUTURA JDISPOSITIVI IMPIANTABILI ATTIVI K DISPOSITIVI PER CHIRURGIA MINI-INVASIVA ED ELETTROCHIRURGIA LSTRUMENTARIO CHIRURGICO PLURIUSO O RIUSABILE MDISPOSITIVI PER MEDICAZIONI GENERALI E SPECIALISTICHE NDISPOSITIVI PER SISTEMA NERVOSO E MIDOLLARE P DISPOSITIVI PROTESICI IMPIANTABILI E PRODOTTI PER OSTEOSINTESI CND (dispositivi medici – ultimo aggiornamento con DM 29.7.2013 )
 FDISPOSITIVI PER DIALISI GDISPOSITIVI PER APPARATO GASTROINTESTINALE HDISPOSITIVI DA SUTURA JDISPOSITIVI IMPIANTABILI ATTIVI K DISPOSITIVI PER CHIRURGIA MINI-INVASIVA ED ELETTROCHIRURGIA LSTRUMENTARIO CHIRURGICO PLURIUSO O RIUSABILE MDISPOSITIVI PER MEDICAZIONI GENERALI E SPECIALISTICHE NDISPOSITIVI PER SISTEMA NERVOSO E MIDOLLARE P DISPOSITIVI PROTESICI IMPIANTABILI E PRODOTTI PER OSTEOSINTESI CND (dispositivi medici – ultimo aggiornamento con DM ).")
14
Q DISPOSITIVI PER ODONTOIATRIA, OFTALMOLOGIA E OTORINOLARINGOIATRIA RDISPOSITIVI PER APPARATO RESPIRATORIO E ANESTESIA SPRODOTTI PER STERILIZZAZIONE T DISPOSITIVI DI PROTEZIONE E AUSILI PER INCONTINENZA (D. Lgs. 46/97) UDISPOSITIVI PER APPARATO UROGENITALE VDISPOSITIVI VARI W DISPOSITIVI MEDICO-DIAGNOSTICI IN VITRO (D. Lgs. 332/2000) YSUPPORTI O AUSILI TECNICI PER PERSONE DISABILI Z APPARECCHIATURE SANITARIE E RELATIVI COMPONENTI ACCESSORI E MATERIALI CND (dispositivi medici – ultimo aggiornamento con DM 29.7.2013 )
 UDISPOSITIVI PER APPARATO UROGENITALE VDISPOSITIVI VARI W DISPOSITIVI MEDICO-DIAGNOSTICI IN VITRO (D. Lgs. 332/2000) YSUPPORTI O AUSILI TECNICI PER PERSONE DISABILI Z APPARECCHIATURE SANITARIE E RELATIVI COMPONENTI ACCESSORI E MATERIALI CND (dispositivi medici – ultimo aggiornamento con DM ).")
15
Es..

18
Numero di repertorio (Decreto 21/12/2009 modifica in parte il Decreto 20/02/2007) Di fatto l‘iscrizione nel Repertorio è obbligatoria per : DM commercializzati in Italia a partire dal 1° maggio 2007 il cui responsabile dell’immissione in commercio abbia sede legale nel territorio italiano L’iscrizione nel Repertorio è facoltativa per: DM di classe I e assemblati commercializzati in Italia a partire dal 1° maggio 2007 il cui responsabile dell’immissione in commercio abbia sede legale al di fuori del territorio italiano DM commercializzati in Italia prima del 1° maggio 2007 Le strutture pubbliche possono acquistare SOLO i dispositivi che sono iscritti nel Repertorio del Ministero della Salute (..se hanno l’obbligo di esserlo..) ….alcuni punti fermi… Tramite il Repertorio si identifica in maniera univoca con un codice numerico ciascun DM. Sul Repertorio Nazionale dei DM (database) è possibile scaricare documentazione relativa a ciascun device.
 è possibile scaricare documentazione relativa a ciascun device..")
19
IL PRONTUARIO DEI DM Grazie al completamento di ogni DM iscritto in Anagrafica Aziendale con CND e Repertorio si è elaborato il Prontuario Aziendale dei DM nel quale sono ricompresi i devices di corrente impiego clinico. Questo risulta di fondamentale importanza al fine di razionalizzare gli acquisti di DM simili o con le medesime indicazioni d’uso, nonché per riconvertire le acquisizioni a procedure di acquisto in gara riducendo in tal senso gli acquisti in economia o in esclusiva.

20
….perché parliamo di gestione e di governo dei DM….?? GESTIONE DEL RISCHIO PER IL PAZIENTE E PER L’OPERATORE GOVERNO DELLE RISORSE 54.000.000€ la spesa per DM della ASL nel 2014 (175.000.000€ spesa complessiva per farmaci ma solo 24.000.000€ quella per farmaci utilizzati in ospedali) Osservanza delle indicazioni d’uso (off-label/in-label) Uso appropriato del DM
 Osservanza delle indicazioni d’uso (off-label/in-label) Uso appropriato del DM.")
21
Come entrano in Azienda i nuovi DM? CADM (Commissione Aziendale Dispositivi Medici) Attività prevalenti: Valuta le richieste nuovi inserimenti Autorizza l’entrata dei DM in prova Analizza i consumi e la spesa di DM innovativi e ad alto impatto economico al fine di migliorare l’appropriatezza d’uso Sviluppa la Dispositivo Vigilanza Le CADM aziendali sono coordinate dalla CRDM che fornisce le linee di indirizzo ed elabora documenti sui DM di particolare complessità. La CRDM funge da interlocutore tra le CADM locali e il Ministero della Salute istituite con Delibera di Giunta Regionale DGR n. 1523/2008
 Attività prevalenti: Valuta le richieste nuovi inserimenti Autorizza l’entrata dei DM in prova Analizza i consumi e la spesa di DM innovativi e ad alto impatto economico al fine di migliorare l’appropriatezza d’uso Sviluppa la Dispositivo Vigilanza Le CADM aziendali sono coordinate dalla CRDM che fornisce le linee di indirizzo ed elabora documenti sui DM di particolare complessità. La CRDM funge da interlocutore tra le CADM locali e il Ministero della Salute istituite con Delibera di Giunta Regionale DGR n. 1523/2008.")
22
1- ITER DI VALUTAZIONE DELLA RICHIESTA DI INTRODUZIONE PER ACQUISIZIONE DI NUOVO DM. A.Compilazione del modulo di richiesta di introduzione da parte del clinico richiedente. B.Validazione della richiesta da parte del Direttore di Dipartimento. C.Stesura dell’istruttoria da parte del servizio di competenza. D. Discussione e decisione della CADM.

23
L’ISTRUTTORIA: FASI PRINCIPALI L’istruttoria viene curata dalla Segreteria Scientifica della CADM ed in particolare dal referente dell’Ingegneria Clinica, dal Farmacista o dal referente del servizio Economale. L’istruttoria consta di varie fasi: raccolta della documentazione tecnica, analisi dei vantaggi attesi, confronto con i DM in uso nella pratica clinica corrente, valutazione di report di HTA e di studi clinici ed infine studio di impatto sul budget assegnato al Servizio che richiede il nuovo DM.

24
2- ITER DI VALUTAZIONE DELLA RICHIESTA DI INTRODUZIONE PER PROVA DI NUOVO DM. Su indicazione della CRDM ogni CADM ha predisposto un percorso per l’introduzione nei servizi aziendali di DM in prova gratuita Compilazione del modulo di richiesta di introduzione per prova da parte del clinico richiedente per i DM appartenenti alle classi di rischio IIb e III. Verifica da parte della CADM: a)conformità del DM alla normativa, b)utilizzo coerente con le indicazioni d’uso del DM; autorizzazione (solo per i DM di classe IIb ) Se il DM è di classe III è prevista l’istruttoria che dovrà riportare le caratteristiche tecniche del DM, l’utilizzo previsto, le motivazioni ecc.. La prova dovrà essere autorizzata sia dalla CADM che dalla DS Aziendale Se il DM è di classe I o IIa non è prevista autorizzazione CADM ma deve essere consegnato attraverso i servizi di Farmacia che registrano il DM in entrata
conformità del DM alla normativa, b)utilizzo coerente con le indicazioni d’uso del DM; autorizzazione (solo per i DM di classe IIb ) Se il DM è di classe III è prevista l’istruttoria che dovrà riportare le caratteristiche tecniche del DM, l’utilizzo previsto, le motivazioni ecc.. La prova dovrà essere autorizzata sia dalla CADM che dalla DS Aziendale Se il DM è di classe I o IIa non è prevista autorizzazione CADM ma deve essere consegnato attraverso i servizi di Farmacia che registrano il DM in entrata.")
25
Le Farmacie registrano, al momento dell’ingresso, il tipo di DM, il nome commerciale, il lotto, la scadenza, le quantità in ingresso, il reparto utilizzatore e il nome del clinico che ne ha richiesto l’uso in prova. Questo percorso permette di conoscere che cosa è stato consegnato e che cosa è presente in giacenza nelle diverse U.U.O.O. aziendali. (Percorso che tutela paziente e operatore) Questo percorso permette: la sicurezza che nei Servizi Aziendali entrino solo DM conformi alla normativa la tracciabilità del DM
 Questo percorso permette: la sicurezza che nei Servizi Aziendali entrino solo DM conformi alla normativa la tracciabilità del DM.")
26
3- MONITORAGGIO E ANALISI DEI CONSUMI; AZIONI PER L'USO APPROPRIATO E IL CONTENIMENTO DEI COSTI Es. Dispositivi medici da monitorare per il Dipartimento di Chirurgia Generale: SUTURATRICI MECCANICHE (utilizzo di prodotti equivalenti a minor costo) EMOSTATICI LOCALI (riviste le indicazioni d'uso – Linee guida RER) TROCAR E DM x ULTRASUONI (utilizzo di prodotti equivalenti a minor costo) RETI CHIRURGICHE (utilizzo di prodotti equivalenti a minor costo) CHIRURGIA ROBOTICA (ridimensionati/bloccati gli acquisti)
 EMOSTATICI LOCALI (riviste le indicazioni d uso – Linee guida RER) TROCAR E DM x ULTRASUONI (utilizzo di prodotti equivalenti a minor costo) RETI CHIRURGICHE (utilizzo di prodotti equivalenti a minor costo) CHIRURGIA ROBOTICA (ridimensionati/bloccati gli acquisti).")
27
Monitoraggio continuo …verifica delle richieste prima dell’acquisto….. Prima dell’emissione degli ordini, sulle richieste che provengono dai servizi/reparti vengono effettuate valutazioni quali-quantitative per verificare che: Il prodotto sia quello che il reparto vuole davvero Che la quantità richiesta sia coerente con l’attività del servizio richiedente Che il prodotto sia quello a contratto; in caso contrario si procede con la sostituzione con quello di gara Che il prodotto richiesto non coperto da contratto sia quello a minor costo tra quelli che sono valutati essere sovrapponibili per utilizzo e caratteristiche

28
Monitoraggio continuo …chi effettua queste valutazioni???..... FARMACIA Dispositivi a gestione ULC ( in carico da sempre) Protesi ed endoprotesi vascolari e cardiache Stent emodinamica Protesi e Endoprotesi vascolari Lentine intraoculari Protesi Mammarie Materiale di Dialisi Protesi esofagee e gastrointestinali Protesi urogenitali SIC Dispositivi collegati alle apparecchiature (Z) DM-IVD (W) per lab.centralizzato-BLU e POCT Robotica Protesi ortopediche Defibrillatori e pacemaker Neurostimolatori Impianti cocleari Tutte le richieste in entrata vengono valutate dai servizi di Farmacia o dal servizio di Ing. Clinica secondo una suddivisione che tiene conto delle rispettive competenze
 Protesi ed endoprotesi vascolari e cardiache Stent emodinamica Protesi e Endoprotesi vascolari Lentine intraoculari Protesi Mammarie Materiale di Dialisi Protesi esofagee e gastrointestinali Protesi urogenitali SIC Dispositivi collegati alle apparecchiature (Z) DM-IVD (W) per lab.centralizzato-BLU e POCT Robotica Protesi ortopediche Defibrillatori e pacemaker Neurostimolatori Impianti cocleari Tutte le richieste in entrata vengono valutate dai servizi di Farmacia o dal servizio di Ing. Clinica secondo una suddivisione che tiene conto delle rispettive competenze.")
29
…quali le modalità CONTRATTUALI…??? Gara d’appalto Trattativa o cottimo fiduciario (solo per importi che non superino i 209.000€/anno; 5 Ditte) Contratto di esclusività (robotica, materiale dedicato, caratteristiche peculiari) Economia (acquisto senza contratto-possibile per importi non sup a 20.000€/anno)
 Contratto di esclusività (robotica, materiale dedicato, caratteristiche peculiari) Economia (acquisto senza contratto-possibile per importi non sup a €/anno).")
30
Gara d’appalto: le principali fasi tecniche Definizione del capitolato tecnico da parte del gruppo di esperti raccolta dei fabbisogni Definizione della scheda di valutazione con i parametri tecnici da valutare e relativi punteggi assegnati Definizione del prezzo a base d’asta Nomina della Commissione di Gara dopo pubblicazione del bando Valutazione del campione di DM proposto dalle Ditte partecipanti e relativa documentazione tecnica Verifica di qualità dei campioni di DM presentati dalle Ditte il cui prodotto è stato ritenuto conforme alla descrizione del lotto dalla Commissione di gara Raccolta delle schede di valutazione Formulazione della graduatoria sulla base del rapporto tra prezzo e offerta (60/40) Aggiudicazione
 Aggiudicazione")
31
Descrizione di un lotto di gara: es. Protesi biologica ad origine suina non cross-linking spessore superiore a 1 mm. In confezione sterile, monouso. Misure, circa 10x15,15x20,20x25 Ditte che hanno un prodotto che potrebbe avere le caratteristiche richieste dal lotto: Ditta A; suino, cross-linking, spessore 1,8mm, misure: 10x15,15x20,20x25 Ditta B; suino, cross-linking, spessore 1,5mm, misure: 10x16,15x20,20x25 Ditta C; suino, cross-linking, spessore 1,3mm, misure: 10x16,16x20,20x25 Ditta D; suino, cross-linking, spessore 1,4mm, misure: 10x15,18x25,20x30,20x10

32
Normativa di riferimento: Linee-guida MEDDEV 2.12-1 rev 8 (2013) D.L. 46/97 emendato con DL 37/10 Decreto Ministeriale 15.11.2005 LA DISPOSITIVO-VIGILANZA NELLA PRATICA CLINICA

33
10 giorni dall’evento (art. 9 D.L. 46/97, comma 1, emendato con D.L 37/10 ) 30 giorni dall’evento (art. 9 D.L. 46/97 emendato con D.L 37/10) Un po’ di definizioni.. MANCATO INCIDENTE Si intende la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonché qualsiasi carenza nell’etichettatura o nelle istruzioni per l’uso di un DM che direttamente o indirettamente AVREBBERO POTUTO CAUSARE un grave peggioramento dello stato di salute o la morte del paziente se il DM fosse stato utilizzato o non fosse intervenuto il personale sanitario INCIDENTE Si intende la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonché qualsiasi carenza nell’etichettatura o nelle istruzioni per l’uso di un DM che direttamente o indirettamente ABBIANO CAUSATO un grave peggioramento dello stato di salute o la morte del paziente
 30 giorni dall’evento (art. 9 D.L. 46/97 emendato con D.L 37/10) Un po’ di definizioni.. MANCATO INCIDENTE Si intende la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonché qualsiasi carenza nell’etichettatura o nelle istruzioni per l’uso di un DM che direttamente o indirettamente AVREBBERO POTUTO CAUSARE un grave peggioramento dello stato di salute o la morte del paziente se il DM fosse stato utilizzato o non fosse intervenuto il personale sanitario INCIDENTE Si intende la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonché qualsiasi carenza nell’etichettatura o nelle istruzioni per l’uso di un DM che direttamente o indirettamente ABBIANO CAUSATO un grave peggioramento dello stato di salute o la morte del paziente.")
34
Cosa si intende per grave peggioramento dello stato di salute? Malattia o lesione con pericolo per la vita Una menomazione di una funzione del corpo o una lesione di una struttura corporea Una condizione che rende necessario un intervento medico o chirurgico per impedire una menomazione di una funzione del corpo o una lesione di una struttura corporea Ospedalizzazione del paziente o prolungamento della stessa Morte del paziente

35
LA DISPOSITIVO VIGILANZA: gli attori coinvolti A livello nazionale chi si occupa della Vigilanza post marketing dei DM è il Ministero della Salute. Il Ministero della Salute ha diversi interlocutori che sono il fabbricante/mandatario e i Responsabili Aziendali della Dispositivo Vigilanza (RAV). Altri protagonisti sono i segnalatori di difettosità a carico dei DM in uso tra cui infermieri, strumentisti e clinici. La Dispositivo Vigilanza si distingue in attiva e passiva che va distinta dal reclamo.
. Altri protagonisti sono i segnalatori di difettosità a carico dei DM in uso tra cui infermieri, strumentisti e clinici. La Dispositivo Vigilanza si distingue in attiva e passiva che va distinta dal reclamo..")
36
LA DISPOSITIVO VIGILANZA ATTIVA Consiste nella segnalazione di incidenti/mancati incidenti rilevati da operatori sanitari durante l’utilizzo di un device. LA DISPOSITIVO VIGILANZA PASSIVA Consiste negli Avvisi di Sicurezza che la Ditta invia al Ministero e agli utilizzatori del device o ritiri da parte del Ministero RECLAMO sono le segnalazioni che si fanno nei casi in cui non vi sia stato un danno effettivo o potenziale per il paziente.

37
normativa Nazionale (art. 9 D.L. 46/97, comma 2, emendato con D.L 37/10) Art.23 D.L. 46/97, comma 1 emendato con D.L. 37/10 1. I fabbricanti o i loro mandatari, gli operatori sanitari, i legali rappresentanti delle strutture sanitarie o, se nominati, i referenti per la vigilanza, che violano le prescrizioni dell’articolo 9, commi 2, 3 o 7, sono puniti con l’arresto fino a sei mesi e con l’ammenda da 7.200 euro a 43.200 euro. Cosa succede se non non si fa…???? Iper-segnalazione……??? Si segnala anche quando non si dovrebbe???? Difficile a volte stabilire se sia o no incidente…..es. confezione di garza che ne contiene un numero inferiore o superiore rispetto al dichiarato. Es. mascherine per anestesia che non si conformano bene al viso… ago cannula, ecc.. ????

38
3. La comunicazione di cui al comma 2 è effettuata direttamente o tramite la struttura sanitaria ove avviene l’incidente segnalato, nel rispetto di eventuali disposizioni regionali che prevedano la presenza di referenti per la vigilanza sui dispositivi medici. normativa Nazionale (art. 9 D.L. 46/97, comma 3, emendato con D.L 37/10) Come si segnala????? L’operatore sanitario può fare la segnalazione: Direttamente con modulistica prevista dal M.d.S. Attraverso referenti (RAV Aziendali) RAV Aziendali
 Come si segnala . L’operatore sanitario può fare la segnalazione: Direttamente con modulistica prevista dal M.d.S. Attraverso referenti (RAV Aziendali) RAV Aziendali.")
39
normativa Nazionale (art. 9 D.L. 46/97, comma 4, emendato con D.L 37/10) A chi si segnala????? La segnalazione va inviata : Al Ministero della Salute (banca dati del Ministero) Al Fabbricante o Mandatario Al Fornitore Alla Regione Emilia Romagna (CRDM) 4. La comunicazione di cui ai commi 2 e 3 deve essere inviata altresì al fabbricante o al suo mandatario, anche per il tramite del fornitore del dispositivo medico.
 Al Fabbricante o Mandatario Al Fornitore Alla Regione Emilia Romagna (CRDM) 4. La comunicazione di cui ai commi 2 e 3 deve essere inviata altresì al fabbricante o al suo mandatario, anche per il tramite del fornitore del dispositivo medico..")
40
normativa Nazionale (art. 9 D.L. 46/97, comma 6, emendato con D.L 37/10) 6. Gli operatori sanitari pubblici o privati sono tenuti a comunicare al fabbricante o al mandatario, direttamente o tramite la struttura sanitaria di appartenenza e, quindi, anche per il tramite del fornitore del dispositivo medico, ogni altro inconveniente che, pur non integrando le caratteristiche dell’incidente di cui al comma 1, lettera a), possa consentire l’adozione delle misure atte a garantire la protezione e la salute dei pazienti e degli utilizzatori. RECLAMO Quando il prodotto non è idoneo ma non può provocare un incidente…??? Si segnala la disfunzione o il difetto direttamente al Fabbricante. Es. capello in confezione sterile, guanti che si rompono…???, sacca urina che si rompe in reparto???
, possa consentire l’adozione delle misure atte a garantire la protezione e la salute dei pazienti e degli utilizzatori. RECLAMO Quando il prodotto non è idoneo ma non può provocare un incidente… . Si segnala la disfunzione o il difetto direttamente al Fabbricante. Es. capello in confezione sterile, guanti che si rompono… , sacca urina che si rompe in reparto .")
41
Un circolo virtuoso Utilizzatore RAV Operatori Sanitari Attori coinvolti

42
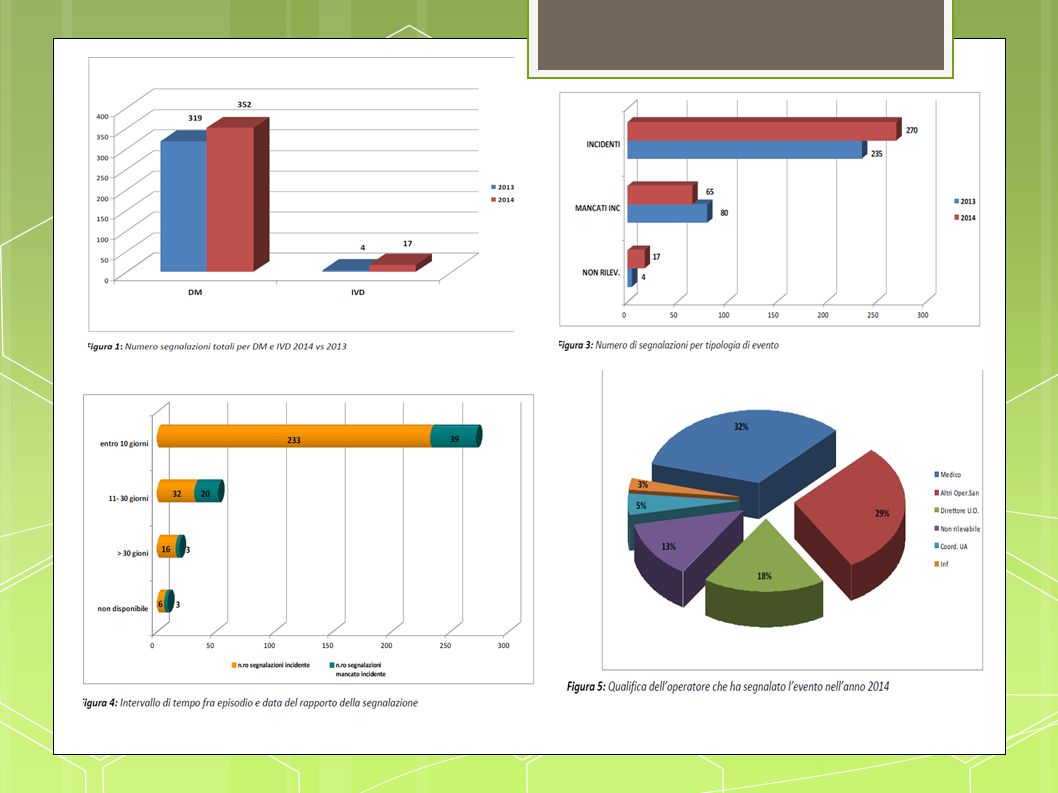
Incidenti-mancati incidenti: Monitoraggio Regionale Anno 2014

44
..es di incidente/mancato incidente... Endo Catch™ Gold sacca di estrazione Ditta Covidien Incidente: rottura di un frammento del cestello durante un intervento di colecistectomia. Recupero del frammento, allungamento dell'intervento chirurgico Azioni del RAV: comunicazione dell'incidente a tutti gli utilizzatori aziendali con indicazione del blocco del lotto. Invio della segnalazione a: Ministero, RER, Fabbricante e Rivenditore

45
..es di avviso di sicurezza... Ritiro da parte della Ditta REDAX del DRENAGGIO PLEURICO CH24 21124 lotto F1504010 Il problema rilevato come descritto nel documento allegato è: Dall'esame dei dati di sorveglianza post-vendita risulta che le unità del lotto indicato presentano un potenziale distaccamento del connettore blu distale del catetere. Rischio potenziale: Il raccordo blu viene normalmente collegato ad un sistema di raccolta dei liquidi. In caso di distacco, si può verificare la disconnessione del drenaggio dal sistema di raccolta con possibilità di infezione del cavo pleurico o possibile sviluppo di pneumotorace.

46
RITIRO DA PARTE DEL M.d.S.

47
..es di segnalazione di reclamo... SIRINGA ML.20 L.F. C/ECC. G20 2020EM38 Ditta RAYS Lotto INJ8811114 Sostituzione dell’intero lotto: se il problema persiste si dichiara l’inidoneità del prodotto e si passa a Ditta alternativa

48
I DISPOSITIVI MEDICI NEGLI STUDI CLINICI: FASE POST MARKET Studi con DM post market In questi casi il device è già in commercio, ha il Marchio CE e viene proposto e utilizzato nello studio secondo le indicazione d’uso contenute nella scheda tecnica del dispositivo e conformi alla marcatura CE. La documentazione dello studio deve contenere: la scheda tecnica del DM le istruzioni d’uso in ITALIANO Il certificato dell’ON che attesta che il DM ha il marchio CE Va verificato: che il DM sia iscritto nel Repertorio Dispositivi Medici del Ministero della Salute Comunicazione dello studio al Ministero Salute Parere favorevole del CE La valutazione clinica svolta dal fabbricante, iniziata nella fase di pre-market, continua dopo la marcatura CE per la conferma della sicurezza e delle prestazioni del dispositivo, dell’accettabilità del rapporto rischio/beneficio e della identificazione di eventuali ulteriori rischi che possono emergere dall’impiego del prodotto su ampia scala e a lungo termine. Studi clinici

49
Es. di studio clinico con DM post-market Studio Osservazionale Post-Approvazione alla commercializzazione (post-market) del dispositivo AMPLATZER™ Amulet™ (Sponsor: St. Jude Medical, Inc) In questo caso il DM: o viene utilizzato secondo l’indicazione d’uso o è già in fornitura con contratto di gara AVEN o lo studio è osservazionale o il DM non viene fornito dallo sponsor D.L.46/97 emendato con DL 37/10 - Art 14 - comma 8 …….indagini cliniche svolte con dispositivi medici recanti la marcatura CE…… Le spese ulteriori rispetto alla normale pratica clinica………… sono a carico del fabbricante. I dispositivi medici occorrenti per le indagini cliniche, che non sono già stati acquisiti nel rispetto delle ordinarie procedure di fornitura dei beni, sono altresì a carico del fabbricante……… Il Dispositivo AMPLTZER Amulet è utilizzato per occludere l’auricola sinistra nei pazienti con fibrillazione atriale Studi clinici
 del dispositivo AMPLATZER™ Amulet™ (Sponsor: St. Jude Medical, Inc) In questo caso il DM: o viene utilizzato secondo l’indicazione d’uso o è già in fornitura con contratto di gara AVEN o lo studio è osservazionale o il DM non viene fornito dallo sponsor D.L.46/97 emendato con DL 37/10 - Art 14 - comma 8 …….indagini cliniche svolte con dispositivi medici recanti la marcatura CE…… Le spese ulteriori rispetto alla normale pratica clinica………… sono a carico del fabbricante. I dispositivi medici occorrenti per le indagini cliniche, che non sono già stati acquisiti nel rispetto delle ordinarie procedure di fornitura dei beni, sono altresì a carico del fabbricante……… Il Dispositivo AMPLTZER Amulet è utilizzato per occludere l’auricola sinistra nei pazienti con fibrillazione atriale Studi clinici.")
50
Es. di studio clinico con DM post-market D.L.46/97 emendato con DL 37/10 - Art 14 - comma 8 …….indagini cliniche svolte con dispositivi medici recanti la marcatura CE…… Le spese ulteriori rispetto alla normale pratica clinica………… sono a carico del fabbricante. I dispositivi medici occorrenti per le indagini cliniche, che non sono già stati acquisiti nel rispetto delle ordinarie procedure di fornitura dei beni, sono altresì a carico del fabbricante……… SISTEMA TREVO RETRIEVER (Sponsor: Stryker Neurovascular) -lo sponsor propone la NON fornitura perché è già in acquisto in ASL In questo caso il DM: o viene utilizzato secondo l’indicazione d’uso o è già in fornitura con contratto di gara AVEN ma in scadenza o È uno studio per valutare l’efficacia e le performance del DM o il DM deve essere fornito dallo sponsor dopo la scadenza del contratto di fornitura DM inserito in un arteria inguinale per il recupero del trombo Studi clinici
 -lo sponsor propone la NON fornitura perché è già in acquisto in ASL In questo caso il DM: o viene utilizzato secondo l’indicazione d’uso o è già in fornitura con contratto di gara AVEN ma in scadenza o È uno studio per valutare l’efficacia e le performance del DM o il DM deve essere fornito dallo sponsor dopo la scadenza del contratto di fornitura DM inserito in un arteria inguinale per il recupero del trombo Studi clinici.")
51
Es. di studio clinico con DM post-market D.L.46/97 emendato con DL 37/10 - Art 14 - comma 8 …….indagini cliniche svolte con dispositivi medici recanti la marcatura CE…… Le spese ulteriori rispetto alla normale pratica clinica………… sono a carico del fabbricante. I dispositivi medici occorrenti per le indagini cliniche, che non sono già stati acquisiti nel rispetto delle ordinarie procedure di fornitura dei beni, sono altresì a carico del fabbricante……… stent INCRAFT® (Sponsor: Cordis Corporation) – lo sponsor propone la NON fornitura del device perché già acquistato In questo caso il DM: o viene utilizzato secondo l’indicazione d’uso o NON è in fornitura; acquistato in esclusiva autorizzata per due casi particolari o È uno studio per valutare l’efficacia e le performance del DM o il DM deve essere fornito dallo sponsor dallo sponsor DM per il trattamento degli aneurismi dell’aorta addominale Studi clinici
 – lo sponsor propone la NON fornitura del device perché già acquistato In questo caso il DM: o viene utilizzato secondo l’indicazione d’uso o NON è in fornitura; acquistato in esclusiva autorizzata per due casi particolari o È uno studio per valutare l’efficacia e le performance del DM o il DM deve essere fornito dallo sponsor dallo sponsor DM per il trattamento degli aneurismi dell’aorta addominale Studi clinici.")
52
o Sicurezza La prima grande sfida è definire, con sempre maggiore accuratezza, quale sia il dispositivo medico più adatto da utilizzare su un determinato paziente anche a fronte del fatto che l’innovazione non sempre si sposa con il concetto di miglior trattamento possibile. o Economicità La seconda sfida sempre più imponente nello scenario odierno è dovuta alla necessità di ottemperare alla appropriatezza clinica sfruttando il giusto grado di innovazione ma disponendo di risorse economiche sempre più limitate. …le sfide nella gestione dei DM….

53
GRAZIE PER L'ATTENZIONE..!

Presentazioni simili