Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
La messa a punto di un procedimento sperimentale di purificazione di una proteina
richiede la possibilità di: 1. Identificare e quantizzare la proteina di interesse fra tutte le altre proteine del campione di partenza (gli ENZIMI possono essere identificati sulla base della reazione che essi catalizzano tramite un opportuno SAGGIO ENZIMATICO) 2. Misurare la quantità di proteine totali nel campione 3. Valutare la presenza di proteine contaminanti nel campione di interesse
 2. Misurare la quantità di proteine totali nel campione. 3. Valutare la presenza di proteine contaminanti nel campione di interesse.")
2
Valore ipotetico proteina pura = 100 U/mg
proteine totali (mg) Enzima (mg) Resa % Purezza (%) U totali As (U/mg) Omogenato 1000 100 100% 10% 10000 10 1° stadio purificazione 5000 2° stadio 2500 3° stadio 1980 Saggio colorimetrico Stima da SDS-PAGE Saggio di attività enzimatica Valore ipotetico proteina pura = 100 U/mg mg o U di prot. di interesse nella frazione mg o U di prot. di interesse all’inizio della purificazione Resa = U di proteina di interesse nella frazione mg di protine totali nella frazione As =
 Enzima (mg) Resa % Purezza (%) U totali. As. (U/mg) Omogenato % 10% ° stadio. purificazione ° stadio ° stadio Saggio. colorimetrico. Stima da SDS-PAGE. Saggio di attività. enzimatica. Valore ipotetico proteina pura = 100 U/mg. mg o U di prot. di interesse nella frazione. mg o U di prot. di interesse all’inizio della purificazione. Resa = U di proteina di interesse nella frazione. mg di protine totali nella frazione. As. =")
3
ELETTROFORESI Tale termine descrive la migrazione di particelle cariche sotto l’influenza di un campo elettrico Molte molecole di interesse biologico possiedono gruppi ionizzabili e, quindi, ad un opportuno valore di pH sono presenti in soluzione come specie elettricamente cariche Sotto l’influenza di un campo elettrico queste molecole cariche migrano verso il catodo o l’anodo, a seconda che possiedano una carica positiva (cationi) o negativa (anioni)
 o negativa (anioni)")
4
PARAMETRI CHIAVE L’elettroforesi è dunque il processo per cui molecole cariche si separano in un campo elettrico a causa delle loro diverse mobilità I fattori che influenzano la mobilità di una molecola in un campo elettrico comprendono la carica della molecola (q), il gradiente di voltaggio del campo elettrico (E), la resistenza di attrito del mezzo di supporto (f) Il prodotto dei parametri q ed E (q x E) fornisce la forza, misurata in newton, che spinge una molecola di carica q verso un elettrodo di carica opposta La forza frizionale f, la quale rallenta il movimento della molecola carica, dipende dalle dimensioni della molecola, dalla sua forma, dalle dimensioni dei pori del mezzo nel quale avviene l’elettroforesi e dalla viscosità del tampone La velocità (v) di una molecola carica che si sposta in un campo elettrico è data dunque dalla seguente equazione: v Eq f
, il gradiente di voltaggio del. campo elettrico (E), la resistenza di attrito del mezzo di supporto (f) Il prodotto dei parametri q ed E (q x E) fornisce la forza, misurata in. newton, che spinge una molecola di carica q verso un elettrodo di carica. opposta. La forza frizionale f, la quale rallenta il movimento della molecola carica, dipende dalle dimensioni della molecola, dalla sua forma, dalle. dimensioni dei pori del mezzo nel quale avviene l’elettroforesi e dalla. viscosità del tampone. La velocità (v) di una molecola carica che si sposta in un campo elettrico. è data dunque dalla seguente equazione: v Eq. f.")
5
MOBILITA' ELETTROFORETICA
Comunemente è adoperato il termine mobilità elettroforetica (μ) dello ione, che equivale alla velocità dello ione diviso l’intensità del campo elettrico (v/E) Quando si applica una differenza di potenziale, molecole con carica elettrica totale differente inizieranno a separarsi in funzione della loro mobilità elettroforetica Anche molecole con carica elettrica uguale, ma con dimensioni molecolari differenti, si separeranno per effetto di forze frizionali diverse Se il campo elettrico è rimosso prima che le varie molecole cariche abbiano raggiunto gli elettrodi, si ha una separazione dei singoli componenti in base alla loro mobilità elettroforetica Una volta terminata la separazione dei campioni, questi sono visualizzati mediante opportuni metodi di colorazione
 dello. ione, che equivale alla velocità dello ione diviso l’intensità del campo. elettrico (v/E) Quando si applica una differenza di potenziale, molecole con carica. elettrica totale differente inizieranno a separarsi in funzione della loro. mobilità elettroforetica. Anche molecole con carica elettrica uguale, ma con dimensioni. molecolari differenti, si separeranno per effetto di forze frizionali. diverse. Se il campo elettrico è rimosso prima che le varie molecole cariche. abbiano raggiunto gli elettrodi, si ha una separazione dei singoli. componenti in base alla loro mobilità elettroforetica. Una volta terminata la separazione dei campioni, questi sono. visualizzati mediante opportuni metodi di colorazione.")
6
Inversamente proporzionale alle DIMENSIONI MOLECOLARI
Elettroforesi - + + + - + catodo anodo Direttamente proporzionale alla CARICA NETTA Velocità di migrazione Inversamente proporzionale alle DIMENSIONI MOLECOLARI

7
Direttamente proporzionale al rapporto
Elettroforesi - + + + - + Velocità di migrazione Direttamente proporzionale al rapporto CARICA / MASSA

8
Elettroforesi su GEL: quando il campo elettrico viene annullato le proteine restano nel punto che avevano raggiunto

9
Elettroforesi su GEL: quando il campo elettrico viene annullato le proteine restano nel punto che avevano raggiunto + -

10
Elettroforesi su GEL: quando il campo elettrico viene annullato le proteine restano nel punto che avevano raggiunto + - Il tampone deve mantenere costante lo stato di ionizzazione delle molecole che devono essere separate

11
Elettroforesi su GEL: quando il campo elettrico viene annullato le proteine restano nel punto che avevano raggiunto Aggiunta del Fissativo

12
MATERIALI DI SUPPORTO Gli studi pionieristici sull’elettroforesi furono condotti in soluzione libera Apparve subito chiaro che i problemi associati a questa metodica, in particolar modo gli effetti negativi della diffusione, potevano essere limitati stabilizzando il mezzo in cui avviene l’elettroforesi Ciò fu realizzato facendo avvenire l’elettroforesi su un supporto meccanico poroso, il quale veniva opportunamente bagnato nel tampone di corsa ed all’interno del quale si realizzava l’elettroforesi degli ioni del tampone e del campione Un mezzo di supporto ideale dovrebbe essere idrofilico (al fine di impedire interazioni idrofobiche tra le molecole del campione ed il mezzo di supporto), privo di carica e stabile in un ampio intervallo di temperatura, pH e osmolarità e dovrebbe avere una porosità controllata e regolabile in modo preciso Oggi per la maggior parte delle tecniche elettroforetiche si adoperano gel di poliacrilammide o di agarosio
, privo di carica e stabile in un ampio intervallo. di temperatura, pH e osmolarità e dovrebbe avere una porosità. controllata e regolabile in modo preciso. Oggi per la maggior parte delle tecniche elettroforetiche si adoperano gel. di poliacrilammide o di agarosio.")
13
POROSITA' DEL MEZZO DI SUPPORTO
Le dimensioni dei pori del mezzo di supporto sono importanti perché contribuiscono al coefficiente di attrito (f) L’effetto di setaccio del mezzo di supporto, insieme al rapporto carica/massa delle molecole, contribuisce a determinare la separazione delle molecole del campione
 L’effetto di setaccio del mezzo di supporto, insieme al. rapporto carica/massa delle molecole, contribuisce a. determinare la separazione delle molecole del campione.")
14
APPARECCHIATURA PER ELETTROFORESI
COMPONENTI PRINCIPALI Alimentatore Cella elettroforetica 1. Per elettroforesi su gel verticale 2. Per elettroforesi su gel orizzontale Se durante una elettroforesi è applicato un voltaggio costante, l’intensità di corrente, per effetto della diminuzione della resistenza, aumenta, determinando un aumento del calore sviluppato (si veda la legge di Ohm: V/I=R). Per tale motivo si utilizzano alimentatori in grado di fornire una corrente costante. In questo modo si eliminano le fluttuazioni di calore.
. Per tale motivo si utilizzano alimentatori in grado di fornire una corrente costante. In questo modo si eliminano le fluttuazioni di calore.")
15
ELETTROFORESI DI PROTEINE
Le proteine hanno una carica netta a qualunque valore di pH diverso dal loro punto isoelettrico (pI) Quando sono poste in un campo elettrico le proteine migreranno verso l’elettrodo di carica opposta Quasi tutte le proteine hanno un pI compreso nell’intervallo 3-10 e la maggioranza di esse ha un pI<8 Ne consegue che ad un pH pari ad 8 o maggiore la maggioranza delle proteine ha una carica netta negativa e migrerà in un campo elettrico verso l’anodo (elettrodo positivo) La tecnica elettroforetica maggiormente adoperata per le molecole proteiche è l’elettroforesi su gel di poliacrilammide in sodio dodecil solfato (SDS-PAGE)
 Quando sono poste in un campo elettrico le proteine migreranno verso. l’elettrodo di carica opposta. Quasi tutte le proteine hanno un pI compreso nell’intervallo 3-10 e la. maggioranza di esse ha un pI<8. Ne consegue che ad un pH pari ad 8 o maggiore la maggioranza delle. proteine ha una carica netta negativa e migrerà in un campo elettrico. verso l’anodo (elettrodo positivo) La tecnica elettroforetica maggiormente adoperata per le molecole. proteiche è l’elettroforesi su gel di poliacrilammide in sodio. dodecil solfato (SDS-PAGE)")
16
La separazione delle molecole cariche sottoposte al campo elettrico si basa essenzialmente sull’effetto setaccio L’SDS-PAGE consente la separazione di molecole con un rapporto carica/massa identico, ma di dimensioni molecolari diverse Nell’SDS-PAGE la matrice del gel è una sostanza reticolata che agisce come un setaccio, in cui le forze di attrito fanno diminuire la mobilità elettroforetica delle molecole in relazione alle loro dimensioni

17
ACRILAMMIDE NORME DI SICUREZZA
Costituisce il mezzo di supporto di scelta per quasi tutte le applicazioni dell’elettroforesi di proteine e di molte applicazioni dell’elettroforesi di acidi nucleici L’acrilammide polimerizzata si presenta sotto forma di gel La porosità del gel di poliacrilammide può essere regolata variando la percentuale di acrilammide e/o il grado di legami trasversali tra le catene di acrilammide NORME DI SICUREZZA L’acrilammide allo stato liquido è una potente neurotossina. Pertanto assicuratevi di indossare i guanti in tutte le operazioni che prevedono l’utilizzo di acrilammide. Quando pesate o utilizzate la polvere, indossate una maschera. Le soluzioni non utilizzate devono essere polimerizzate prima di essere eliminate. Ogni liquido versato deve essere assorbito immediatamente con tovaglioli di carta ed i tovaglioli eliminati in un recipiente apposito per rifiuti tossici. Una volta polimerizzata l’acrilammide perde tossicità.

18
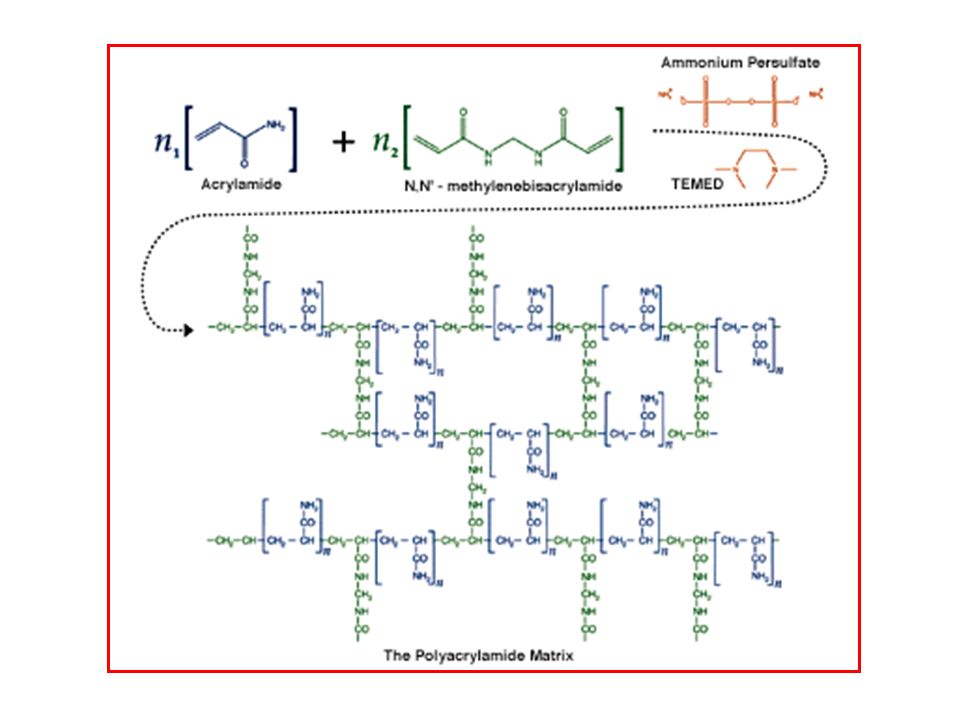
POLIMERIZZAZIONE I gel di poliacrilammide sono preparati facendo copolimerizzare monomeri di acrilammide in presenza di piccole quantità di N,N’-metilene bisacrilammide (bis-acrilamide). La bis-acrilammide, composta da due molecole di acrilammide legate da un gruppo metilene, è utilizzata come agente in grado di formare legami crociati (cross-linking agent). I monomeri di acrilammide polimerizzano nel senso testa-coda e occasionalmente si legano ad una molecola di bis-acrilammide. In tal modo nella catena è introdotto un secondo sito per l’estensione. Ciò fa sì che si formi una matrice con dei legami crociati a struttura ben definita. Acrilammide Bis-acrilammide
. La bis-acrilammide, composta da due molecole di acrilammide. legate da un gruppo metilene, è utilizzata come agente in grado. di formare legami crociati (cross-linking agent). I monomeri di acrilammide polimerizzano nel senso testa-coda e. occasionalmente si legano ad una molecola di bis-acrilammide. In tal modo nella catena è introdotto un secondo sito per. l’estensione. Ciò fa sì che si formi una matrice con dei legami. crociati a struttura ben definita. Acrilammide. Bis-acrilammide.")
19
Il processo di polimerizzazione dell’acrilammide è un tipico esempio
di catalisi radicalica ed inizia con l’aggiunta di ammonio persolfato e della base N,N,N’,N’-tetrametilendiammina (TEMED). Il TEMED catalizza la decomposizione dello ione persolfato con la produzione del corrispondente radicale libero (cioè una molecola con un elettrone spaiato), nel modo seguente: Se rappresentiamo il radicale libero con R· ed il monomero di acrilammide con M, possiamo schematizzare la polimerizzazione nel modo seguente: R· + M → RM· RM· + M → RMM· RMM· + M → RMMM· ecc. L’ammonio persolfato è l’estere disolfato dell’acqua ossigenata e omolisa rapidamente a radicali instabili (radicali solforici). Il TEMED è un’ammina terziaria che reagisce con questi radicali a formare radicali liberi TEMED, che a loro volta reagiscono con l’acrilammide inducendone la polimerizzazione. TEMED (iniziatore)
. Il TEMED catalizza la decomposizione dello ione persolfato con la. produzione del corrispondente radicale libero (cioè una molecola. con un elettrone spaiato), nel modo seguente: Se rappresentiamo il radicale libero con R· ed il monomero di. acrilammide con M, possiamo schematizzare la polimerizzazione nel. modo seguente: R· + M → RM· RM· + M → RMM· RMM· + M → RMMM· ecc. L’ammonio persolfato. è l’estere disolfato. dell’acqua ossigenata e. omolisa rapidamente a. radicali instabili. (radicali solforici). Il TEMED è un’ammina. terziaria che reagisce. con questi radicali a. formare radicali liberi. TEMED, che a loro volta. reagiscono con. l’acrilammide inducendone. la polimerizzazione. TEMED (iniziatore)")
20
In questo modo si formano lunghe catene di acrilammide tenute
insieme tra loro da legami crociati derivanti dall’inserzione occasionale all’interno della catena di molecole di bis-acrilammide. Dal momento che l’ossigeno rimuove i radicali liberi dalla soluzione, tutte le soluzioni per la preparazione del gel sono degassate prima dell’uso. Le soluzioni sono poste in beute da vuoto e poste per breve tempo sotto vuoto per allontanare l’ossigeno disciolto. Acrilammide bis-acrilammide (catalizzatore) TEMED (iniziatore) GEL DI POLIACRILAMMIDE (molto idrofilico => trattiene grosse quantità di acqua)
 TEMED (iniziatore) GEL DI POLIACRILAMMIDE. (molto idrofilico => trattiene grosse quantità di acqua)")
21
FOTOPOLIMERIZZAZIONE
E’ un metodo alternativo per la polimerizzazione dei gel di acrilammide. In questo caso al posto dell’ammonio persolfato e del TEMED si utilizza riboflavina. La soluzione è illuminata per 2 o 3 ore con una luce intensa. La conseguente fotodecomposizione della riboflavina produce radicali liberi che danno inizio alla polimerizzazione del gel.

24
Le dimensioni medie dei pori in un gel di poliacrilammide possono essere
controllate variando la quantità di monomero (acrilammide) o aumentando il grado di legami trasversali per ottenere pori più stretti. In genere il grado di legami trasversali è mantenuto costante, mentre si varia la percentuale di acrilammide per ottenere gel di differente porosità. Per un gel di una data composizione si avrà una distribuzione statistica di dimensioni dei pori. Proteine relativamente piccole migreranno nel gel con un impedimento minimo, mentre la migrazione delle proteine di dimensioni maggiori sarà ritardata. In ogni dato gel saranno dunque separate solo le proteine che sono in un particolare ambito di dimensioni
 o aumentando. il grado di legami trasversali per ottenere pori più stretti. In genere il grado di legami trasversali è mantenuto costante, mentre si. varia la percentuale di acrilammide per ottenere gel di differente porosità. Per un gel di una data composizione si avrà una distribuzione statistica di. dimensioni dei pori. Proteine relativamente piccole migreranno nel gel con un impedimento. minimo, mentre la migrazione delle proteine di dimensioni maggiori sarà. ritardata. In ogni dato gel saranno dunque separate solo le proteine che sono in un. particolare ambito di dimensioni.")
25
Percentuale di acrilammide Ambito ottimale di dimensioni
molecolari (Da) 5-12 10-15 >15 <15.000 N.B.: la percentuale di bis-acrilammide è fissa e corrisponde a circa il 5% dell’acrilammide.
 >15. < N.B.: la percentuale di bis-acrilammide è fissa e corrisponde a circa il 5% dell’acrilammide.")
26
SDS-PAGE Per semplificare l’analisi di miscele di proteine è possibile fare in modo che la separazione elettroforetica si basi solo sulle dimensioni delle catene polipeptidiche. Ciò si ottiene denaturando le proteine con il detergente Sodio Dodecil Solfato (SDS). Tale detergente si lega con forza alle proteine, le quali si ripiegano a formare delle bacchette estese rivestite di SDS. In media, ogni due amminoacidi sarà presente una molecola di SDS. Poiché ciascuna molecola di SDS ha due cariche negative ai valori di pH adoperati per l’elettroforesi, la carica netta delle catene polipeptidiche rivestite sarà molto più negativa di quella delle catene non rivestite. Inoltre il rapporto carica/massa sarà essenzialmente identico per proteine diverse, dal momento che il rivestimento di SDS domina la carica. La separazione delle catene polipeptidiche denaturate e rivestite di detergente si baserà quasi esclusivamente sulle dimensioni delle molecole proteiche
. Tale detergente si lega con forza alle proteine, le quali si. ripiegano a formare delle bacchette estese rivestite di SDS. In media, ogni due amminoacidi sarà presente una molecola di SDS. Poiché ciascuna molecola di SDS ha due cariche negative ai valori di pH. adoperati per l’elettroforesi, la carica netta delle catene polipeptidiche. rivestite sarà molto più negativa di quella delle catene non rivestite. Inoltre il rapporto carica/massa sarà essenzialmente identico per. proteine diverse, dal momento che il rivestimento di SDS domina la. carica. La separazione delle catene polipeptidiche. denaturate e rivestite di detergente. si baserà quasi esclusivamente. sulle dimensioni delle molecole proteiche.")
27
Sodio Dodecil Solfato CH3(CH2)10CH2OSO3- Na+ proteina
10CH2OSO3- Na+ proteina")
28
Per ottenere una buona separazione di proteine diverse in una miscela è
essenziale che le proteine siano applicate sul gel in volumi molto piccoli La parte superiore del gel di poliacrilammide, nota come stacking gel, è versata direttamente al di sopra del resolving gel Lo stacking gel ha delle proprietà che consentono la concentrazione delle proteine del campione in una zona sottile al di sopra del resolving gel In tal modo le proteine rivestite da SDS sono separate nel resolving gel in maniera efficace e riproducibile Lo stacking gel è polimerizzato con una piccola percentuale di acrilammide e di bis-acrilammide, per assicurare un’alta porosità, ed è tamponato con tampone Tris-HCl a pH 6,8 Il resolving gel contiene invece una percentuale più alta di acrilammide ed è tamponato con Tris-HCl a pH 8,8 Il tampone di corsa contiene Tris a pH 8,3 con glicina come controione

29
PRINCIPI DELLA COMPRESSIONE
Quando la glicina del tampone di corsa entra nello stacking gel a pH 6,8 sarà principalmente nella sua forma zwitterionica neutra, con una piccola frazione (1%) in forma di ione glicinato carico negativamente Ciò impedisce alla glicina di essere un trasportatore efficiente di corrente Gli ioni Cl- restano trasportatori efficienti di corrente a pH 6,8 e migrano rapidamente verso l’anodo Durante questa elettroforesi nello stacking gel, la concentrazione degli ioni Cl- scende drasticamente all’estremità catodica del gel, formando un gradiente crescente di concentrazione verso l’anodo Le molecole proteiche rivestite di SDS ed il colorante, i quali hanno rapporti carica/massa maggiori di quello della glicina ma minori di quello del Cl-, devono adesso migrare per portare la corrente di elettroforesi dietro gli ioni Cl- e davanti alla glicina A mano a mano che procede l’elettroforesi, le molecole proteiche che raggiungono il resolving gel sono ritardate notevolmente, consentendo alle molecole proteiche che le seguono di raggiungerle Il volume del campione di proteine “presentato” al gel di risoluzione sarà molto più piccolo del volume caricato inizialmente sullo stacking gel
 in forma di ione glicinato carico negativamente. Ciò impedisce alla glicina di essere un trasportatore efficiente di corrente. Gli ioni Cl- restano trasportatori efficienti di corrente a pH 6,8 e migrano rapidamente verso l’anodo. Durante questa elettroforesi nello stacking gel, la concentrazione degli ioni Cl- scende drasticamente all’estremità catodica del gel, formando un gradiente crescente di concentrazione verso l’anodo. Le molecole proteiche rivestite di SDS ed il colorante, i quali hanno rapporti carica/massa maggiori di quello della glicina ma minori di quello del Cl-, devono adesso migrare per portare la corrente di elettroforesi dietro gli ioni Cl- e davanti alla glicina. A mano a mano che procede l’elettroforesi, le molecole proteiche che raggiungono il resolving gel sono ritardate notevolmente, consentendo alle molecole proteiche che le seguono di raggiungerle. Il volume del campione di proteine presentato al gel di risoluzione sarà molto più piccolo del volume caricato inizialmente sullo stacking gel.")
30
RICORDARSI CHE... Quando viene applicata la corrente, tutte le specie ioniche presenti devono migrare alla stessa velocità altrimenti si verifica una interruzione nel circuito elettrico. Perché ciò avvenga è necessario che gli ioni glicinato, più lenti, siano soggetti ad un campo elettrico più intenso rispetto agli ioni Cl- più veloci. Il campo elettrico è inversamente proporzionale alla conduttività, la quale a sua volta è proporzionale alla concentrazione dello ione che conduce corrente. Il risultato è che le tre specie ioniche tendono a variare la propria concentrazione in modo che gli ioni Cl- siano più concentrati dei complessi SDS-proteina, i quali, a loro volta, siano più concentrati del glicinato. Dal momento che la quantità di complessi SDS-proteina è molto inferiore alla quantità di ioni glicinato, questi complessi tendono a concentrarsi in una banda molto sottile tra il glicinato e gli ioni Cl-.

31
PRINCIPI DELLA SEPARAZIONE
Quando il glicinato raggiunge il gel di separazione, per effetto del pH più alto (6,8 nello stacking gel e 8,8 nel resolving gel), diviene completamente ionizzato e quindi aumenta la propria mobilità Dunque nel gel di separazione gli ioni glicinato e gli ioni Cl- migrano più velocemente rispetto ai complessi SDS-proteina, i quali migrano in maniera inversamente proporzionale alle loro dimensioni molecolari La separazione dei complessi SDS-proteina avviene infatti in base agli effetti di setaccio molecolare dovuti alle dimensioni dei pori del gel Le proteine più piccole passano più facilmente all’interno dei pori del gel, mentre le proteine di dimensioni maggiori sono ritardate dalle forze frizionali
, diviene completamente ionizzato. e quindi aumenta la propria mobilità. Dunque nel gel di separazione gli ioni glicinato e gli ioni Cl- migrano più. velocemente rispetto ai complessi SDS-proteina, i quali migrano in maniera. inversamente proporzionale alle loro dimensioni molecolari. La separazione dei complessi SDS-proteina avviene infatti in base agli effetti di. setaccio molecolare dovuti alle dimensioni dei pori del gel. Le proteine più piccole passano più facilmente all’interno dei pori del gel, mentre. le proteine di dimensioni maggiori sono ritardate dalle forze frizionali.")
Presentazioni simili







![N2O4 2 NO2 d[N2O4] d[NO2] V = - = 1/ 2 dt dt V = k [N2O4]n k, costante specifica di velocità, aumenta all’aumentare della temperatura. n, ordine.](/1/553488/big_thumb.jpg "N2O4 2 NO2 d[N2O4] d[NO2] V = - = 1/ 2 dt dt V = k [N2O4]n k, costante specifica di velocità, aumenta all’aumentare della temperatura. n, ordine.>")








polimeri è quello della determinazione del peso molecolare. I polimeri.>")
 sottoposte a un campo elelttrico.>")


