Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
TECNICHE DI CAMPIONAMENTO E MISURA DEGLI INQUINANTI ATMOSFERICI
Università degli Studi di Napoli Federico II Facoltà di Ingegneria Corso di Monitoraggio di Inquinanti nell’Ambiente

2
Indice dei composti analizzati
Biossido di zolfo Ossidi di azoto Monossido di carbonio Benzene Ozono Particolato

3
BIOSSIDO DI ZOLFO – SO2 Principio fisico:
Metodo di riferimento normalizzato per la misurazione della concentrazione di biossido di zolfo: UNI EN 14212:2005 Metodo di misura: fluorescenza ultravioletta Principio fisico: Le molecole di SO2 attivate per irraggiamento con radiazioni ultraviolette rilasciano energia per fluorescenza La intensità della energia rilasciata per fluorescenza viene messa in correlazione con la concentrazione di SO2 nel campione

4
Fenomeno della fluorescenza
Eccitazione da S0 a S2 per assorbimento di radiazione UV Rilascio di energia (calore) fino a , stato vibrazionale fondamentale (v=0) dello stato elettronico superiore (S1) Transizione radiativa a partire dallo stato vibrazionale fondamentale (v=0) dello stato elettronico superiore (S1) verso stati vibrazionali dello stato elettronico fondamentale (S0) Diagramma di Jablonski L’energia irradiata per fluorescenza ha una lunghezza d’onda maggiore di quella assorbita
 fino a , stato vibrazionale fondamentale (v=0) dello stato elettronico superiore (S1) Transizione radiativa a partire dallo stato vibrazionale fondamentale (v=0) dello stato elettronico superiore (S1) verso stati vibrazionali dello stato elettronico fondamentale (S0) Diagramma di Jablonski. L’energia irradiata per fluorescenza ha una lunghezza d’onda maggiore di quella assorbita.")
5
Meccanismo di fluorescenza per la SO2
(1) SO2+ hν1 → SO2* (2) SO2* → SO2+hν2 Il meccanismo è il seguente (1) La molecola di SO2 assorbe la radiazione UV a frequenza v1 (214 nm) e passa alla stato eccitato SO2* (2) SO2* ritorna nello stato molecolare rilasciando l’eccesso di energia hν2 ad una differente lunghezza d’onda (tra 240 e 420 nm centrata a 330 nm)
 SO2+ hν1 → SO2* (2) SO2* → SO2+hν2. Il meccanismo è il seguente. (1) La molecola di SO2 assorbe la radiazione UV a frequenza v1 (214 nm) e passa alla stato eccitato SO2* (2) SO2* ritorna nello stato molecolare rilasciando l’eccesso di energia hν2 ad una differente lunghezza d’onda. (tra 240 e 420 nm centrata a 330 nm)")
6
Schema analizzatore SO2
1) L'aria ambiente viene prelevata in continuo e viene inviata in una cella di misura 2) La cella viene irradiata da una radiazione UV resa monocromatica da un filtro; 3) Le molecole di SO2 eccitate emettono una radiazione specifica a lunghezza d'onda più grande. 4) Un tubo foto moltiplicatore (PMT) rileva la radiazione emessa dal decadimento di SO2 * produce un segnale analogico in output
 L aria ambiente viene prelevata in continuo e viene inviata in una cella di misura. 2) La cella viene irradiata da una radiazione UV resa monocromatica da un filtro; 3) Le molecole di SO2 eccitate emettono una radiazione specifica a lunghezza d onda più grande. 4) Un tubo foto moltiplicatore (PMT) rileva la radiazione emessa. dal decadimento di SO2 * produce un segnale analogico in output.")
7
Schema analizzatore SO2
La radiazione UV è generata da una lampada che produce la quantità massima di luce alla lunghezza d'onda necessaria per eccitare SO2 in SO2 * (214 nm) Un circuito rivelatore di riferimento misura costantemente l’intensità della lampada in modo da poter compensare il guadagno dell’amplificatore di segnale in funzione del decadimento dell’energia emessa dalla lampada stessa. Per assicurare inoltre che il PMT riveli soltanto la luce emessa dal decadimento di SO2 * (330 nm) il percorso di eccitazione della luce UV e il campo di visibilità di PMT sono tra loro perpendicolari
 Un circuito rivelatore di riferimento misura costantemente l’intensità della lampada in modo da poter compensare il guadagno dell’amplificatore di segnale in funzione del decadimento dell’energia emessa dalla lampada stessa. Per assicurare inoltre che il PMT riveli soltanto la luce emessa dal decadimento di SO2 * (330 nm) il percorso di eccitazione della luce UV e il campo di visibilità di PMT sono tra loro perpendicolari.")
9
Interferenze e quenching
Il meccanismo completo è il seguente: (1) SO2 + hv1 → SO2* (2) SO2* → SO2 + hν2 (3) SO2* + hv2 → SO + (O) (4) SO2*+ M → SO2+ M I processi (3) e (4) sono in competizione con il processo di fluorescenza (2). (3) fotolisi: SO2* si decompone per effetto della luce emessa dalle molecole nello stato eccitato. (4) quenching: le molecole nello stato eccitato perdono energia nella collisione con altre molecole (principalmente O2 e H2O). Il disturbo del fenomeno di quenching sulle misure può essere minimizzato calibrando l’analizzatore con la stessa miscela che contiene la specie da analizzare. Nel caso di misure in aria ambiente, l’analizzatore viene calibrato con SO2 in aria. In tal modo c’è la stessa concentrazione di O2 (composto M) nella miscela di calibrazione e nella miscela da analizzare.
 SO2 + hv1 → SO2* (2) SO2* → SO2 + hν2 (3) SO2* + hv2 → SO + (O) (4) SO2*+ M → SO2+ M. I processi (3) e (4) sono in competizione con il processo di fluorescenza (2). (3) fotolisi: SO2* si decompone per effetto della luce emessa dalle molecole nello stato eccitato. (4) quenching: le molecole nello stato eccitato perdono energia nella collisione con altre molecole (principalmente O2 e H2O). Il disturbo del fenomeno di quenching sulle misure può essere minimizzato calibrando l’analizzatore con la stessa miscela che contiene la specie da analizzare. Nel caso di misure in aria ambiente, l’analizzatore viene calibrato con SO2 in aria. In tal modo c’è la stessa concentrazione di O2 (composto M) nella miscela di calibrazione e nella miscela da analizzare.")
10
Calcolo della concentrazione di SO2
La legge di Lambert Beer stabilisce che l’energia assorbita da un mezzo attraversato da una radiazione I0 è data da : Ia = I0 [ 1− exp( − a L CSO2)] dove: Ia = Intensità della radiazione assorbita I0 = Intensità della luce UV di eccitazione a = coefficiente di assorbimento del mezzo (SO2) L = Distanza tra la sorgente UV e le molecole SO2 che vengono influenzate (lunghezza percorso o cammino ottico) CSO2 = Concentrazione di SO2 nella camera di misura
] dove: Ia = Intensità della radiazione assorbita. I0 = Intensità della luce UV di eccitazione. a = coefficiente di assorbimento del mezzo (SO2) L = Distanza tra la sorgente UV e le molecole SO2 che vengono influenzate (lunghezza percorso o cammino ottico) CSO2 = Concentrazione di SO2 nella camera di misura.")
11
Calcolo della concentrazione di SO2
La intensità della radiazione dovuta al decadimento di SO2* dipende dalla velocità con cui questa reazione avviene (k costante cinetica). F = k (SO2 *) dove: F = Intensità della radiazione di luce fluorescente k = velocità con cui SO2* decade a SO2 SO2* = quantità di SO2 eccitato nella camera campione la intensità della radiazione di fluorescenza (F) dipende quindi da: - quantità di SO2* eccitato velocità di decadimento (k) Ia = I0 [ 1− exp (− a L CSO2)] - concentrazione di SO2; - intensità della luce UV (I0); - lunghezza del percorso della luce (L) temperatura
. F = k (SO2 *) dove: F = Intensità della radiazione di luce fluorescente. k = velocità con cui SO2* decade a SO2. SO2* = quantità di SO2 eccitato nella camera campione. la intensità della radiazione di fluorescenza (F) dipende quindi da: - quantità di SO2* eccitato. velocità di decadimento (k) Ia = I0 [ 1− exp (− a L CSO2)] - concentrazione di SO2; - intensità della luce UV (I0); - lunghezza del percorso della luce (L) temperatura.")
12
Calcolo della concentrazione di SO2
Se - l'intensità della luce (I0) è conosciuta e costante; - la lunghezza del percorso della luce eccitata è breve e costante (L); - la temperatura del gas è costante o conosciuta e compensata (k costante); - minimizzazione o costanza delle condizioni interferenti presenti. la quantità (F) emessa di luce fluorescente è direttamente collegata con la concentrazione di SO2 nella camera campione.
 è conosciuta e costante; - la lunghezza del percorso della luce eccitata è breve e costante (L); - la temperatura del gas è costante o conosciuta e compensata (k costante); - minimizzazione o costanza delle condizioni interferenti presenti. la quantità (F) emessa di luce fluorescente è direttamente collegata con la concentrazione di SO2 nella camera campione.")
13
INTERFERENZA POSITIVA
L’analizzatore di SO2 misura ogni specie che assorbe a circa nm e fluoresce a nm. Misure sperimentali hanno individuato tra queste l’ossido di azoto (NO), il disolfuro di carbonio (CS2) e l’etilene (C2H2). Fluorescenza di SO2 ed NO. Il rapporto tra la fluorescenza di SO2 ed NO aiuta nella scelta della zona da rivelare per minimizzare l’interferenza dell’NO. Spettro di assorbimento di SO2 e NO nella regione nm.
, il disolfuro di carbonio (CS2) e l’etilene (C2H2). Fluorescenza di SO2 ed NO. Il rapporto tra la fluorescenza di SO2 ed NO aiuta nella scelta della zona da rivelare per minimizzare l’interferenza dell’NO. Spettro di assorbimento di SO2 e NO nella regione nm.")
14
OSSIDI DI AZOTO - NOx E’ un fenomeno analogo alla fluorescenza
Metodo di riferimento normalizzato per la misurazione della concentrazione di biossido di azoto ed ossidi di azoto: ISO 7996:1985 Pricipio di misura: chemiluminescenza E’ un fenomeno analogo alla fluorescenza L’eccitazione è dovuta a una reazione chimica

15
CNO2 = C (NO+NO2) - CNO Principio di funzionamento
L’analizzatore misura la concentrazione di monossido di azoto che reagendo con ozono produce NO2 eccitato: NO + O3 -> NO2* + O2 Le molecole di NO2 eccitate elettronicamente decadono a stati energetici inferiori rilasciando energia sotto forma di radiazione luminosa rossa nella banda compresa tra 0.6 e 3 micron. NO2* -> NO2 + hv Per misurare il biossido di azoto questo deve essere trasformato in monossido attraverso un convertitore catalitico al molibdeno a 375 °C: 3 NO2 + Mo = 3 NO + MoO3 CNO2 = C (NO+NO2) - CNO
 - CNO.")
16
Schema analizzatore NOx

17
Aspetti tecnologici analizzatore NOx
1) L’ozono è prodotto all’interno dello strumento dall’ossigeno contenuto dell’aria, per mezzo di una scarica ad alta tensione. 2) L’emissione di luce è filtrata per eliminare l’interferenza di altri gas quali CO, SO2, idrocarburi insaturi. Essa è misurata da un foto moltiplicatore. 3) Il segnale in uscita è proporzionale alla concentrazione nel campione. 4) La concentrazione di NO2 viene calcolata per differenza tra la concentrazione di NOx totali e la concentrazione di NO. Quest’ultima è ovviamente misurata facendo reagire il campione gassoso direttamente con ozono, senza farlo passare nel convertitore di NO2. In tal modo solo l’NO contenuto nella miscela gassosa viene misurato. Gli strumenti CLD offrono un vasto range di misura con tempi di risposta veloci. NO2 to NO converter Campione da analizzare Al detector CLD NOx NO
 L’ozono è prodotto all’interno dello strumento dall’ossigeno contenuto dell’aria, per mezzo di una scarica ad alta tensione. 2) L’emissione di luce è filtrata per eliminare l’interferenza di altri gas quali CO, SO2, idrocarburi insaturi. Essa è misurata da un foto moltiplicatore. 3) Il segnale in uscita è proporzionale alla concentrazione nel campione. 4) La concentrazione di NO2 viene calcolata per differenza tra la concentrazione di NOx totali e la concentrazione di NO. Quest’ultima è ovviamente misurata facendo reagire il campione gassoso direttamente con ozono, senza farlo passare nel convertitore di NO2. In tal modo solo l’NO contenuto nella miscela gassosa viene misurato. Gli strumenti CLD offrono un vasto range di misura con tempi di risposta veloci. NO2 to NO converter. Campione da analizzare. Al detector CLD. NOx. NO.")
18
MONOSSIDO DI CARBONIO - CO
METODO UNI EN 14626: Metodo normalizzato per la misurazione della concentrazione di monossido di carbonio mediante spettroscopia a raggi infrarossi non dispersiva Principio di funzionamento assorbimento selettivo della radiazione infrarossa in una finestra molto stretta di lunghezze d’onda (4.5 – 5 mm). le molecole di CO assorbono la radiazione infrarossa convertendo l’energia luminosa in energia rotazionale o vibrazionale delle molecole, rilevabile sotto forma di calore.
. le molecole di CO assorbono la radiazione infrarossa convertendo l’energia luminosa in energia rotazionale o vibrazionale delle molecole, rilevabile sotto forma di calore.")
19
Schema analizzatore CO

20
Funzionamento analizzatore CO
Gli emettitori infrarossi (E) sono filamenti riscaldati. Il rivelatore (D) è un volume di gas identico a quello da misurare (nel nostro caso CO). La maggior parte degli strumenti usano due fasci paralleli generati da due fonti infrarosse identiche. I fasci infrarossi sono modulati da un selettore rotante (M). Un fascio attraversa una cella di assorbimento (A) che contiene il gas da analizzare. L'altro fascio attraversa una cella di riferimento (R) riempita di gas inerte, come azoto. Dopo essere passati attraverso una cella di compensazione (F) i due fasci attraversano due camere riempite di gas da misurare che costituiscono un sensore differenziale di pressione (D). Esso consiste di due alloggiamenti identici separati da un diaframma (C) che si muove fra le piastre di un condensatore. La differenza nel calore ricevuto in ogni camera dovuta alla presenza del gas da analizzare sul percorso di uno dei due fasci, causa l’aumento di pressione su un lato del diaframma. La radiazione che ha attraversato la camera di riferimento, infatti, riscalda la cella del detector corrispondente, causando la deformazione della membrana. Il segnale in uscita (S) dalla cella rappresenta le variazioni della capacità del sistema. In alcuni strumenti, le due camere del rivelatore sono collegate da un vaso capillare per compensare i gradienti della temperatura ambiente. Poiché l’analizzatore NDIR è sensibile alla pressione, questo parametro deve rimanere costante durante la calibratura e l'analisi. È inoltre sensibile al vapore acqueo. I materiali utilizzati in alcuni analizzatori IR sono inoltre sensibili ad acqua. I campioni del gas per analisi IR sono quindi freddi e deumidificati.
 sono filamenti riscaldati. Il rivelatore (D) è un volume di gas identico a quello da misurare (nel nostro caso CO). La maggior parte degli strumenti usano due fasci paralleli generati da due fonti infrarosse identiche. I fasci infrarossi sono modulati da un selettore rotante (M). Un fascio attraversa una cella di assorbimento (A) che contiene il gas da analizzare. L altro fascio attraversa una cella di riferimento (R) riempita di gas inerte, come azoto. Dopo essere passati attraverso una cella di compensazione (F) i due fasci attraversano due camere riempite di gas da misurare che costituiscono un sensore differenziale di pressione (D). Esso consiste di due alloggiamenti identici separati da un diaframma (C) che si muove fra le piastre di un condensatore. La differenza nel calore ricevuto in ogni camera dovuta alla presenza del gas da analizzare sul percorso di uno dei due fasci, causa l’aumento di pressione su un lato del diaframma. La radiazione che ha attraversato la camera di riferimento, infatti, riscalda la cella del detector corrispondente, causando la deformazione della membrana. Il segnale in uscita (S) dalla cella rappresenta le variazioni della capacità del sistema. In alcuni strumenti, le due camere del rivelatore sono collegate da un vaso capillare per compensare i gradienti della temperatura ambiente. Poiché l’analizzatore NDIR è sensibile alla pressione, questo parametro deve rimanere costante durante la calibratura e l analisi. È inoltre sensibile al vapore acqueo. I materiali utilizzati in alcuni analizzatori IR sono inoltre sensibili ad acqua. I campioni del gas per analisi IR sono quindi freddi e deumidificati.")
21
BENZENE – C6H6 Metodo di misura UNI EN 14662-1-5:2005
Metodo normalizzato per la misurazione delle concentrazioni di benzene Parte 1: Campionamento per pompaggio seguito da desorbimento termico e gascromatografia Parte 2: Campionamento per pompaggio seguito da desorbimento con solvente e gascromatografia Parte 3: Campionamento per pompaggio automatizzato con gascromatografia in situ Parte 4: Campionamento diffusivo seguito da desorbimento termico e gascromatografia Parte 5: Campionamento diffusivo seguito da desorbimento con solvente e gascromatografia

22
CAMPIONAMENTO PASSIVO/DIFFUSIVO
BENZENE – C6H6 CAMPIONAMENTO PASSIVO/DIFFUSIVO 1 Diffusione attraverso parete porosa 2 Adsorbimento su cartuccia CAMPIONATORE PASSIVO RADIALE

23
Non si utilizzano pompe né occorrono allacciamenti elettrici
BENZENE – C6H6 Campionatori passivi Non si utilizzano pompe né occorrono allacciamenti elettrici Una cartuccia di materiale adsorbente (carbone attivo)determina un gradiente di concentrazione Il benzene diffonde per diffusione attraverso la parte cilindrica porosa La concentrazione in aria si ottiene valutando la massa di benzene adsorbito e il tempo di esposizione attraverso formule fornite dalla casa costruttrice utile per la misura di C medie su tempi di esposizione lunghi > 24 ore
determina un gradiente di concentrazione. Il benzene diffonde per diffusione attraverso la parte cilindrica porosa. La concentrazione in aria si ottiene valutando la massa di benzene adsorbito e il tempo di esposizione attraverso formule fornite dalla casa costruttrice. utile per la misura di C medie su. tempi di esposizione lunghi > 24 ore.")
24
BENZENE – C6H6 CAMPIONAMENTO ATTIVO - ASPIRAZIONE ARIA CON POMPA
ADSORBIMENTO SU CARTUCCIA La cartuccia di campionamento è costituita da un tubicino di vetro (lunghezza 150mm, diametro esterno 6mm, diametro interno 3mm), parzialmente riempita con materiale adsorbente. La fase di campionamento si effettua a flusso costante ( condizioni operative: 50 mL/min per 5 minuti).
, parzialmente riempita con materiale adsorbente. La fase di campionamento si effettua a flusso costante ( condizioni operative: 50 mL/min per 5 minuti).")
25
Analisi Benzene L’analisi viene effettuata dopo il campionamento e prevede 1 - Desorbimento 2 - Analisi di gas cromatografia METODI DI DESORBIMENTO - DESORBIMENTO CHIMICO consiste in una estrazione con solvente, che estrae le sostanze da analizzare adsorbite dalla cartuccia; - DESORBIMENTO TERMICO La cartuccia è desorbita portando rapidamente la temperatura fino a 250°C, sotto regime di flusso di elio (l5mL/min per 6min) in controcorrente rispetto al verso di caricamento del benzene. Il benzene è termicamente desorbito e trasferito alla colonna capillare gascromatografica usata per la separazione e analisi, per mezzo del gas di trasporto (elio).
 in controcorrente rispetto al verso di caricamento del benzene. Il benzene è termicamente desorbito e trasferito alla colonna capillare gascromatografica usata per la separazione e analisi, per mezzo del gas di trasporto (elio).")
26
Cromatografia Fase stazionaria Fase mobile
La cromatografia é una tecnica di separazione di vari componenti di una miscela Sfrutta la diversa attitudine che ogni molecola o ione possiede nel distribuirsi tra due differenti fasi (una stazionaria e una mobile) per adsorbimento. La fase stazionaria può essere costituita da un solido o un liquido opportunamente supportato; la fase mobile é costituita da un fluido liquido o gassoso. Fase stazionaria Una sostanza introdotta in un sistema a due fasi si distribuirà fra le diverse fasi a seconda delle sue particolari proprietà chimico-fisiche. Fase mobile Indicando con Cm e Cs le sue concentrazioni nella fase mobile e nella fase stazionaria rispettivamente, e supponendo il raggiungimento di condizioni di equilibrio, il coefficiente di distribuzione K è pari a: K = Cs / Cm
 per adsorbimento. La fase stazionaria può essere costituita da un solido o un liquido opportunamente supportato; la fase mobile é costituita da un fluido liquido o gassoso. Fase stazionaria. Una sostanza introdotta in un sistema a due fasi si distribuirà fra le diverse fasi a seconda delle sue particolari proprietà chimico-fisiche. Fase mobile. Indicando con Cm e Cs le sue concentrazioni nella fase mobile e nella fase stazionaria rispettivamente, e supponendo il raggiungimento di condizioni di equilibrio, il coefficiente di distribuzione K è pari a: K = Cs / Cm.")
27
Realizzazione della separazione
1 Si posizione la fase stazionaria all’interno di un tubo (colonna) 2 Si fa scorrere nel tubo una fase fluida (eluente) 3 Si inietta nella fase liquida un volume della soluzione da analizzare 4 La soluzione si muove lungo il tubo insieme all’eluente che viene alimentato in continuo 5 Le diverse specie chimiche impiegano tempi diversi (tempo di ritenzione) ad attraversare il tubo a seconda della maggiore o minore affinità nei confronti della fase stazionaria 6 All’uscita della colonna un rivelatore registra l’uscita della specie generando il cromatogramma
 2 Si fa scorrere nel tubo una fase fluida (eluente) 3 Si inietta nella fase liquida un volume della soluzione da analizzare. 4 La soluzione si muove lungo il tubo insieme all’eluente che viene alimentato in continuo. 5 Le diverse specie chimiche impiegano tempi diversi (tempo di ritenzione) ad attraversare il tubo a seconda della maggiore o minore affinità nei confronti della fase stazionaria. 6 All’uscita della colonna un rivelatore registra l’uscita della specie generando il cromatogramma.")
28
Rappresentazione della separazione cromatografica
Cromatogramma

29
Tempo di eluizione e coefficiente di distribuzione
Isoterma di distribuzione di due sostanze A e B B è più affine alla fase stazionaria di A KB > KA A uscirà prima di B dalla colonna Una specie che non ha alcuna affinità con la fase stazionaria avrà lo stesso tempo di eluizione dell’eluente t = S x L / Q S = sezione della colonna cromatografica L = lunghezza colonna Q = portata dell’eluente N.B. trattandosi di una relazione di equilibrio dipende da T Variando opportunamente la T si può favorire la separazione

30
Esempio di cromatogramma di miscela di pesticidi
Risposta analizzatore, massa o concentrazione Esempio di cromatogramma di miscela di pesticidi Linea di zero Picchi cromatografici tempo

31
Meccanismi chimico-fisici della separazione in cromatografia
Adsorbimento: La fase stazionaria é un solido sulla cui superficie si trovano dei siti attivi in grado di stabilire legami secondari (dipolo-dipolo, ponte di idrogeno, Van der Waals) con le diverse molecole della miscela da risolvere. Se la fase mobile é un liquido si parla di cromatografia liquido-solido (LSC), se invece é un gas, di cromatografia gas-solido (GSC). In genere, le molecole che più facilmente vengono fissate sono quelle che presentano gruppi polari, anche se la natura dell’adsorbente influisce sul fenomeno. L’aumento di temperatura agisce negativamente sull’adsorbimento in quanto provoca una maggior agitazione termica. Ripartizione: La fase stazionaria é un liquido, in cui si verifica una vera e propria solubilizzazione delle sostanze da analizzare. Esse pertanto si ripartiscono fra le due fasi (immiscibili tra loro) e la costante K prende il nome di coefficiente di ripartizione e la legge K = Cs / Cm legge di Nernst. Se la fase mobile é un gas si parla di cromatografia gas-liquido (GLC), se invece é un liquido, di cromatografia liquido-liquido (LLC). Scambio ionico: La fase stazionaria é costituita da molecole contenenti gruppi attivi, dotati di cariche elettriche (positive o negative), i quali sono in grado di scambiare i propri ioni con la soluzione da cui vengono lambiti, attraverso un meccanismo di competizione tra gli ioni della fase stazionaria e quelli con la stessa carica contenuti nella fase mobile. Esclusione: Utilizzando una fase solida porosa (o un gel) con pori di opportune dimensioni, è possibile rallentare maggiormente le particelle più piccole che, penetrando nei pori, vengono poi trattenute.
 con le diverse molecole della miscela da risolvere. Se la fase mobile é un liquido si parla di cromatografia liquido-solido (LSC), se invece é un gas, di cromatografia gas-solido (GSC). In genere, le molecole che più facilmente vengono fissate sono quelle che presentano gruppi polari, anche se la natura dell’adsorbente influisce sul fenomeno. L’aumento di temperatura agisce negativamente sull’adsorbimento in quanto provoca una maggior agitazione termica. Ripartizione: La fase stazionaria é un liquido, in cui si verifica una vera e propria solubilizzazione delle sostanze da analizzare. Esse pertanto si ripartiscono fra le due fasi (immiscibili tra loro) e la costante K prende il nome di coefficiente di ripartizione e la legge K = Cs / Cm legge di Nernst. Se la fase mobile é un gas si parla di cromatografia gas-liquido (GLC), se invece é un liquido, di cromatografia liquido-liquido (LLC). Scambio ionico: La fase stazionaria é costituita da molecole contenenti gruppi attivi, dotati di cariche elettriche (positive o negative), i quali sono in grado di scambiare i propri ioni con la soluzione da cui vengono lambiti, attraverso un meccanismo di competizione tra gli ioni della fase stazionaria e quelli con la stessa carica contenuti nella fase mobile. Esclusione: Utilizzando una fase solida porosa (o un gel) con pori di opportune dimensioni, è possibile rallentare maggiormente le particelle più piccole che, penetrando nei pori, vengono poi trattenute.")
32
CROMATOGRAFIA LIQUIDA
Classificazione dei metodi cromatografici La classificazione fondamentale dei metodi cromatografici si basa sullo stato fisico della fase mobile: GAS LIQUIDO GAS CROMATOGRAFIA CROMATOGRAFIA LIQUIDA

33
Tecniche cromatografiche
Cromatografia su strato sottile (TLC): Cromatografia su carta (PC) Cromatografia su colonna a bassa pressione (LPC) Cromatografia in fase liquida ad elevate prestazioni (HPLC) Cromatografia liquida Colonne impaccate Colonne capillari Gas Cromatografia
: Cromatografia su carta (PC) Cromatografia su colonna a bassa pressione (LPC) Cromatografia in fase liquida ad elevate prestazioni (HPLC) Cromatografia liquida. Colonne impaccate. Colonne capillari. Gas Cromatografia.")
34
Tecniche cromatografiche
Cromatografia su strato sottile (TLC): la fase stazionaria può essere gel di silice, allumina, cellulosa in polvere, fatta aderire ad un apposito supporto (alluminio, carta plastificata, lastra di vetro) e la fase mobile é costituita da vari solventi organici. Cromatografia su carta (PC): la fase stazionaria é costituita dall’acqua inevitabilmente presente nella cellulosa come umidità (20%), anche se la carta può essere all’occorrenza trattata con liquidi diversi, e la fase mobile é scelta in funzione del tipo di fase stazionaria e delle proprietà chimiche dei composti da separare. Quasi sempre comunque é una miscela contenente acqua. Cromatografia su colonna a bassa pressione (LPC): La fase mobile é un liquido organico a bassa viscosità mentre le fasi stazionarie, solide, liquide o gel, possono avere caratteristiche chimico-fisiche molto variabili. La tecnica prevede la deposizione in testa ad una colonna (impaccata con un’opportuna fase fissa) di una certa quantità di miscela da separare. Facendo scorrere l’eluente lungo questa colonna si ottiene una certa distribuzione dei componenti della miscela lungo la fase stazionaria. Cromatografia in fase liquida ad elevate prestazioni (HPLC): consiste nella versione strumentale della cromatografia su colonna. L'eluente viene fatto fluire ad alta pressione e le sostanze in uscita vengono rilevate strumentalmente con opportuni dispositivi. Gascromatografia (GC): la fase mobile è un gas che fluisce attraverso una colonna in cui si trova la fase stazionaria solida granulare porosa oppure un liquida. L’unica limitazione della gas-cromatografia é la necessità di rendere volatili i campioni da analizzare.
: la fase stazionaria può essere gel di silice, allumina, cellulosa. in polvere, fatta aderire ad un apposito supporto (alluminio, carta plastificata, lastra di vetro) e la fase mobile. é costituita da vari solventi organici. Cromatografia su carta (PC): la fase stazionaria é costituita dall’acqua inevitabilmente presente nella cellulosa come umidità (20%), anche se la carta può essere all’occorrenza trattata con liquidi diversi, e la fase mobile é scelta in funzione del tipo di fase stazionaria e delle proprietà chimiche dei composti da separare. Quasi sempre comunque é una miscela contenente acqua. Cromatografia su colonna a bassa pressione (LPC): La fase mobile é un liquido organico a bassa viscosità mentre le fasi stazionarie, solide, liquide o gel, possono avere caratteristiche chimico-fisiche molto variabili. La tecnica prevede la deposizione in testa ad una colonna (impaccata con un’opportuna fase fissa) di una certa quantità di miscela da separare. Facendo scorrere l’eluente lungo questa colonna si ottiene una certa distribuzione dei componenti della miscela lungo la fase stazionaria. Cromatografia in fase liquida ad elevate prestazioni (HPLC): consiste nella versione strumentale della cromatografia su colonna. L eluente viene fatto fluire ad alta pressione e le sostanze in uscita vengono rilevate strumentalmente con opportuni dispositivi. Gascromatografia (GC): la fase mobile è un gas che fluisce attraverso una colonna in cui si trova la fase stazionaria solida granulare porosa oppure un liquida. L’unica limitazione della gas-cromatografia é la necessità di rendere volatili i campioni da analizzare.")
35
Schema di un gascromatografo
Componenti principali 1 iniettore 2 colonna 3 camera termostata (forno) 4 rivelatore Altri componenti bombole gas carrier sistema di iniezione (siringa) bombole gas detector software eluente
 4 rivelatore. Altri componenti. bombole gas carrier. sistema di iniezione (siringa) bombole gas detector. software. eluente.")
36
Funzioni dei diversi componenti un gc
gas di trasporto (carrier) può essere (azoto, elio, argon), Trasporta i componenti della miscela in analisi lungo la colonna cromatografica iniettore o camera di iniezione Assicurare l’istantanea vaporizzazione del campione colonna Separazione dei componenti la miscela camera termostatica (forno) Controllo della temperatura ottimale per la separazione modalità isoterma o a temperatura programmata Rivelare l’uscita della colonna di una sostanza diversa dall’eluente rivelatore o detector sistema di iniezione Garantisce l’iniezione istantanea del campione Elabora il segnale in uscita dal rivelatore, costruisce le curve di calibrazione, fornisce i cromatogrammi e le concentrazioni delle specie individuate software
 può essere (azoto, elio, argon), Trasporta i componenti della miscela in analisi lungo la colonna cromatografica. iniettore o camera di iniezione. Assicurare l’istantanea vaporizzazione del campione. colonna. Separazione dei componenti la miscela. camera termostatica (forno) Controllo della temperatura ottimale per la separazione modalità isoterma o a temperatura programmata. Rivelare l’uscita della colonna di una sostanza diversa dall’eluente. rivelatore o detector. sistema di iniezione. Garantisce l’iniezione istantanea del campione. Elabora il segnale in uscita dal rivelatore, costruisce le curve di calibrazione, fornisce i cromatogrammi e le concentrazioni delle specie individuate. software.")
37
Iniettore ingresso carrier iniettore in vetro colonna Siringhe per gc
L’iniettore o camera di iniezione ha il compito di assicurare l’istantanea vaporizzazione del campione. Poiché con l’uso di colonne capillari la quantità di campione da iniettare é dell’ordine dei nanolitri, sono state messe a punto particolari tecniche di iniezione (SPLIT) che consentono di far entrare effettivamente in colonna solo una parte (ad esempio ca. 1/100) del liquido iniettato. La camera di iniezione è corredata da un sistema di resistenze variabili attraverso le quali è possibile fissare la temperatura ritenuta più adatta per la vaporizzazione della miscela. ingresso carrier Camera di vaporizzazione iniettore in vetro colonna Siringhe per gc Volume iniettato ≈ μL
 che consentono di far entrare effettivamente in colonna solo una parte (ad esempio ca. 1/100) del liquido iniettato. La camera di iniezione è corredata da un sistema di resistenze variabili attraverso le quali è possibile fissare la temperatura ritenuta più adatta per la vaporizzazione della miscela. ingresso carrier. Camera di vaporizzazione. iniettore in vetro. colonna. Siringhe per gc. Volume iniettato ≈ μL.")
38
Colonne impaccate per gc
Materiale: tubi di teflon, acciaio o vetro borosilicato disattivato Dimensioni: diametro 2-5 mm lunghezza metri Fase stazionaria: costituita da un solido di supporto ed un liquido non volatile. Il solido di supporto è spesso gel di silice, allumina o carbone, che viene impregnato del liquido che costituisce l'effettiva fase stazionaria. La scelta del liquido è in funzione dei composti che si vogliono separare. In genere si usa squalene, olio o grasso di silicone, glicoli polietilenici (Carbowax), oli di vaselina o trietanolammina, ma la scelta è estremamente ampia.
, oli di vaselina o trietanolammina, ma la scelta è estremamente ampia.")
39
Colonne capillari per gc
Materiale: quarzo Dimensioni: diametro interno 0,1-0,8 mm, lunghezza m Fase stazionaria: film di spessore costante 0.5 – 2.5 micron WCOT (Wall Coated Open Tubular) la fase stazionarie aderisce direttamente alla parete del tubo SCOT (Supported Coated Open Tubular) la fase stazionarie è ancorata d un supporto La fase stazionaria è spesso costituita da metil-silossani (siliconi) con opportuni gruppi funzionali Le diverse fasi stazinarie si differenziano per la polarità: apolari , poco polari, polari
 la fase stazionarie aderisce direttamente alla parete del tubo. SCOT (Supported Coated Open Tubular) la fase stazionarie è ancorata d un supporto. La fase stazionaria è spesso costituita da metil-silossani (siliconi) con opportuni gruppi funzionali. Le diverse fasi stazinarie si differenziano per la polarità: apolari , poco polari, polari.")
40
Camera termostata (forno)
Permette di controllare la temperatura della colonna e quindi la ripartizione delle specie tra fase stazionaria e fase mobile Intervalli di T - T amb 300 – 400 °C - T < 0°C - Analisi isoterme - Analisi con programma d temperatura T[°C] 120 100 Esempio di programma di temperatura per forno GC Ramp rate 2°C/min 40 10 15 20 30 40 Ramp rate 12°C/min tempo [min]

41
Rivelatori Universali Individuano tutti i componenti di una miscela
Selettivi Individuano solo alcune famiglie di composti Distruttivi Modificano i composti eluiti Non distruttivi Non modificano i composti eluiti (disponibili per un’analisi successiva)
")
42
Rivelatore a ionizzazione di fiamma (FID)
Rivelatore universale ma distruttivo in quanto i campioni vengono bruciati per ottenerne la trasformazione in ioni allo stato gassoso. Il rivelatore è costituito da una fiamma di idrogeno e aria alimentati ad un ugello dove arriva anche il gas carrier in uscita dalla colonna La fiamma risultante dalla combustione di idrogeno in aria pirolizza i composti organici producendo atomi caricati positivamente (cationi) ed elettroni. I cationi prodotti dall'elevato calore della fiamma vengono attratti dall'elettrodo negativo ricco di elettroni. Nel momento dell'incontro del catione con l'elettrodo negativo, questi gli cede gli elettroni mancanti generando una debole corrente tra i due elettrodi. La corrente viene rilevata tramite un sensibile amperometro e quindi visualizzata su di un display. Se il carrier gas trasporta dei composti organici la fiamma provoca la pirolisi intermedia dei soluti organici con formazione di radicali R. e HC.. Questi ultimi reagiscono con i radicali O. presenti nella fiamma, secondo la reazione: HC. + O HCO+ + e- SI FORMANO CATIONI ED ELETTRONI
 ed elettroni. I cationi prodotti dall elevato calore della fiamma vengono attratti dall elettrodo negativo ricco di elettroni. Nel momento dell incontro del catione con l elettrodo negativo, questi gli cede gli elettroni mancanti generando una debole corrente tra i due elettrodi. La corrente viene rilevata tramite un sensibile amperometro e quindi visualizzata su di un display. Se il carrier gas trasporta dei composti organici la fiamma provoca la pirolisi intermedia dei soluti organici con formazione di radicali R. e HC.. Questi ultimi reagiscono con i radicali O. presenti nella fiamma, secondo la reazione: HC. + O . HCO+ + e- SI FORMANO CATIONI ED ELETTRONI.")
43
Rivelatore a ionizzazione di fiamma (FID)
- Tra l’ugello stesso della fiamma e il cilindro metallico che lo circonda (catodo -) viene applicata una d.d.p. di circa 300 volt. In modo che il primo funga da anodo (+) e il secondo da catodo (-) La fiamma risultante dalla combustione di idrogeno in aria pirolizza i composti organici producendo atomi caricati positivamente (cationi) ed elettroni. I cationi prodotti dall'elevato calore della fiamma vengono attratti dall'elettrodo negativo ricco di elettroni. Nel momento dell'incontro del catione con l'elettrodo negativo, questi gli cede gli elettroni mancanti generando una debole corrente tra i due elettrodi. La corrente viene rilevata tramite un sensibile amperometro e quindi visualizzata su di un display. + Gli ioni positivi migrano verso il catodo e gli elettroni verso l’anodo con produzione di una corrente elettrica capace di fornire un segnale. La risposta del FID non dipende solo dalla concentrazione del soluto, ma dal numero di atomi di carbonio presenti nella molecola (clorurati rispondono male) Il rivelatore FID quindi registra una intensità di corrente (mA) La sensibilità è elevata ≈ 1 ng
 viene applicata una d.d.p. di circa 300 volt. In modo che il primo funga da anodo (+) e il secondo da catodo (-) La fiamma risultante dalla combustione di idrogeno in aria pirolizza i composti organici producendo atomi caricati positivamente (cationi) ed elettroni. I cationi prodotti dall elevato calore della fiamma vengono attratti dall elettrodo negativo ricco di elettroni. Nel momento dell incontro del catione con l elettrodo negativo, questi gli cede gli elettroni mancanti generando una debole corrente tra i due elettrodi. La corrente viene rilevata tramite un sensibile amperometro e quindi visualizzata su di un display. + Gli ioni positivi migrano verso il catodo e gli elettroni verso l’anodo con produzione di una corrente elettrica capace di fornire un segnale. La risposta del FID non dipende solo dalla concentrazione del soluto, ma dal numero di atomi di carbonio presenti nella molecola (clorurati rispondono male) Il rivelatore FID quindi registra una intensità di corrente (mA) La sensibilità è elevata ≈ 1 ng.")
44
CARATTERISTICHE DEL FID
selettività: universale (composti organici) sensibilità: g = 1 pg (risposta proporzionale al numero di C) stabilità: elevata modalità: distruttiva gas di trasporto: N2 , He temperatura limite: < 400°C esigenze: carrier gas puri e flusso costante I limiti del FID sono la sua scarsa sensibilità verso i gruppi funzionali C=O, -NH2, -OH e l’impossibilità di ottenere il segnale elettrico dalle molecole che non possono essere carbonizzate (He, Ne, Ar, Kr, Xe, O2, N2, H2O, CS2, H2S, SO2, NO, N2O, NO2, NH3, CO, CO2, SiCl4, SiF4). FLUSSI IMPACCATE CAPILLARI carrier gas ml/min ml/min idrogeno 30 ml/min ml/min aria 600 ml/min 350 ml/min
 sensibilità: 10-12g = 1 pg (risposta proporzionale al numero di C) stabilità: elevata. modalità: distruttiva. gas di trasporto: N2 , He. temperatura limite: < 400°C. esigenze: carrier gas puri e flusso costante. I limiti del FID sono la sua scarsa sensibilità verso i gruppi funzionali C=O, -NH2, -OH e l’impossibilità di ottenere il segnale elettrico dalle molecole che non possono essere carbonizzate (He, Ne, Ar, Kr, Xe, O2, N2, H2O, CS2, H2S, SO2, NO, N2O, NO2, NH3, CO, CO2, SiCl4, SiF4). FLUSSI IMPACCATE CAPILLARI. carrier gas ml/min 1-2 ml/min. idrogeno 30 ml/min 25 ml/min. aria 600 ml/min 350 ml/min.")
45
Rivelatore a termoconducibilità (HWD)
Rivelatore universale e non distruttivo Principio di funzionamento un gas, fluendo su un filamento riscaldato, ne abbassa la temperatura rimuovendo parte del calore. La quantità di calore rimosso dipende dal flusso e dalla conducibilità termica del gas che fluisce. In una miscela di gas, pertanto, dipende dalla conducibilità termica di ogni componente (soluti, solvente, gas di trasporto) e dalla pressione parziale di ciascuno di essi. cella di misura filamenti cella di riferimento
 e dalla pressione parziale di ciascuno di essi. cella di. misura. filamenti. cella di. riferimento.")
46
Rivelatore a termoconducibilità (TCD o HWD hot wire)
Il corpo del TCD a doppia cella è un blocco metallico di massa ed inerzia termica elevate nel quale sono ricavate due celle. Esso è termostatato ad una temperatura di circa 50°C superiore a quella massima della colonna, in modo da evitare fenomeni di condensazione. cella di misura Nelle due celle è alloggiato un filamento di tungsteno (o di leghe tungsteno/renio) riscaldato elettricamente. filamenti La cella di riferimento è attraversata dal carrier gas La cella di misure dall’effluente dalla colonna (carrier gas, solventi e soluti) cella di riferimento
 riscaldato elettricamente. filamenti. La cella di riferimento è attraversata dal carrier gas. La cella di misure dall’effluente dalla colonna (carrier gas, solventi e soluti) cella di. riferimento.")
47
Rivelatore a termoconducibilità (HWD)
La resistenza R (in ohm) del filamento varia in funzione della temperatura R1 = R0 [1+0,04 x (T1-T0)] I gas di trasporto impiegati nel TCD sono quelli a più elevata conducibilità termica (H2 e He, preferito per problemi di sicurezza e di minore reattività con i soluti) La presenza di soluti produce un abbassamento della conducibilità termica del carrier gas quindi un aumento di temperatura del filamento che determina un aumento della sua resistenza R, ovvero una diminuzione dell’intensità I della corrente. cella di misura filamenti Le variazioni di conducibilità si misurano indirettamente con il ponte di Wheatstone, un circuito a sei lati e quattro nodi idoneo alla misura di precisione delle resistenze medie cella di riferimento
 del filamento varia in funzione della temperatura. R1 = R0 [1+0,04 x (T1-T0)] I gas di trasporto impiegati nel TCD sono quelli a più elevata conducibilità termica (H2 e He, preferito per problemi di sicurezza e di minore reattività con i soluti) La presenza di soluti produce un abbassamento della conducibilità termica del carrier gas quindi un aumento di temperatura del filamento che determina un aumento della sua resistenza R, ovvero una diminuzione dell’intensità I della corrente. cella di. misura. filamenti. Le variazioni di conducibilità si misurano indirettamente con il ponte di Wheatstone, un circuito a sei lati e quattro nodi idoneo alla misura di precisione delle resistenze medie. cella di. riferimento.")
48
Esempio di cromatogramma da rivelatore a termoconducibilità (HWD)
CH4 CO O2 N2 H2 Le variazioni di conducibilità si misurano indirettamente con il ponte di Wheatstone, un circuito a sei lati e quattro nodi idoneo alla misura di precisione delle resistenze medie

49
CARATTERISTICHE DEL TCD
selettività: universale sensibilità: g (10 ng) stabilità: buona modalità: non distruttiva gas di trasporto: He temperatura limite: < 450°C esigenze: flusso e temperatura costanti
 stabilità: buona. modalità: non distruttiva. gas di trasporto: He. temperatura limite: < 450°C. esigenze: flusso e temperatura costanti.")
50
ELECTRON CAPTURE DETECTOR (ECD)
Il rivelatore a cattura di elettroni si basa sulla ionizzazione primaria delle sostanze per emissione di particelle b- (elettroni veloci) da parte di un debole emettitore, come l’isotopo radioattivo 63Ni. Quando nel detector entra solo il carrier gas (ad esempio Ar) avviene la ionizzazione con formazione di un plasma di elettroni e cationi Ar+, a cui segue la loro migrazione (accelerati da un d.d.p. tra gli elettrodi) rispettivamente all’anodo e al catodo. Si genera quindi una corrente elettrica stazionaria all’interno del circuito di misura. ANODO + 63Ni 63 Ni placca di oro d.d.p. 63Ni 63Ni + b Ar e CATODO - 50
 da parte di un debole emettitore, come l’isotopo radioattivo 63Ni. Quando nel detector entra solo il carrier gas (ad esempio Ar) avviene la ionizzazione con formazione di un plasma di elettroni e cationi Ar+, a cui segue la loro migrazione (accelerati da un d.d.p. tra gli elettrodi) rispettivamente all’anodo e al catodo. Si genera quindi una corrente elettrica stazionaria all’interno del circuito di misura. ANODO + 63Ni. 63 Ni. placca di oro. d.d.p. 63Ni. 63Ni. + b. Ar. e. CATODO")
51
Quando nel detector entra il carrier gas con i componenti del campione separati dalla colonna, se i soluti contengono gruppi elettron-attrattori (alogeni, perossidi, chinoni, nitrogruppi, ecc.), avviene il fenomeno inverso, ovvero una ionizzazione secondaria dei gruppi elettron-attrattori in seguito alla cattura di una frazione dei numerosi elettroni presenti nel circuito di misura. La conseguenza è una diminuzione della corrente che è, ovviamente, il segnale rivelatore del soluto. Gli anioni che si formano hanno una mobilità ridotta verso l’anodo (rispetto quella degli elettroni verso lo stesso anodo) e dunque una conducibilità elettrica talmente modesta da non alterare il valore della diminuzione di corrente elettrica dovuta alla cattura degli elettroni. e ANODO + - d.d.p. R-Cl CATODO - 51

52
L’ECD è costituiito da un corpo con pareti di piombo, non permeabile alle radiazioni b. L’elettrodo centrale cilindrico (anodo +) è circondato da un cilindro cavo la cui parete interna è in parte rivestita da una placca di oro sulla quale è stato elettrodepositato il 63Ni, la parete esterna del rivelatore è l’altro elettrodo (catodo -). Tra i due elettrodi è applicata una d.d.p. che genera una corrente elettrica costante (D.C.). elettrodo centrale (ANODO) + pareti di Pb d.d.p.(D.C.) 63Ni cilindro cavo elettrodo esterno (CATODO) 52
 + pareti di Pb. d.d.p.(D.C.) 63Ni. cilindro cavo. elettrodo esterno. (CATODO) 52.")
53
CARATTERISTICHE DELL’ECD
selettività: elevata sensibilità: g = 1 pg range dinamico: 103 stabilità: moderata modalità: non distruttiva gas di trasporto: N2 o Ar (+ 5% CH4) temperatura limite: < 350°C esigenze: carrier gas purissimo 53
 temperatura limite: < 350°C. esigenze: carrier gas purissimo. 53.")
54
Rivelatore a spettrometria di massa (MS)
Tecnica distruttiva basata sulla ionizzazione di una molecola e sulla sua successiva frammentazione in ioni di diverso rapporto massa / carica (M/z). Il principio su cui si basa è il seguente: una molecola è ionizzata per espulsione di un elettrone; il catione radicalico che si forma (ione molecolare) in parte si frammenta dando molecole e/o radicali neutri (che lo strumento non rileva), in parte generando cationi e/o radicali cationi (ioni frammento). Lo ione molecolare e i vari ioni che si originano per frammentazione (cationi e radicali cationi) vengono discriminati sulla base del loro rapporto massa/carica e rivelati da un detector. Il risultato è lo spettro di massa, che rappresenta l’abbondanza relativa degli ioni in funzione del loro rapporto massa/carica. Questa tecnica consente di misurare le masse molecolari e di ottenere dei profili di frammentazione che sono specifici per ciascun composto.
. Il principio su cui si basa è il seguente: una molecola è ionizzata per espulsione di un elettrone; il catione radicalico che si forma (ione molecolare) in parte si frammenta dando molecole e/o radicali neutri (che lo strumento non rileva), in parte generando cationi e/o radicali cationi (ioni frammento). Lo ione molecolare e i vari ioni che si originano per frammentazione (cationi e radicali cationi) vengono discriminati sulla base del loro rapporto massa/carica e rivelati da un detector. Il risultato è lo spettro di massa, che rappresenta l’abbondanza relativa degli ioni in funzione del loro rapporto massa/carica. Questa tecnica consente di misurare le masse molecolari e di ottenere dei profili di frammentazione che sono specifici per ciascun composto.")
55
Rivelatore a spettrometria di massa (MS)
Tecnica distruttiva basata sulla ionizzazione di una molecola e sulla sua successiva frammentazione in ioni di diverso rapporto massa / carica (M/z). Principio di funzionamento: una molecola è ionizzata per espulsione di un elettrone Si forma il catione radicalico (ione molecolare) e frammenti cationi e/o radicali cationi (ioni frammento). Questi vengono discriminati sulla base del loro rapporto massa/carica e rivelati da un detector.
. Principio di funzionamento: una molecola è ionizzata per espulsione di un elettrone. Si forma il catione radicalico (ione molecolare) e frammenti cationi e/o radicali cationi (ioni frammento). Questi vengono discriminati sulla base del loro rapporto massa/carica e rivelati da un detector.")
58
Schema di uno spettrometro di massa
Il vuoto (che si aggira intorno ai 10-6 – 10-5 torr) è necessario per impedire una perdita di ionizzazione per urto con i gas atmosferici. Introduzione del campione L’introduzione del campione nella camera di ionizzazione può essere fatta sia allo stato solido, usando una sonda, che allo stato liquido o gassoso, usando un sistema di valvole che permettono di accedere alla camera di ionizzazione senza che questa venga a contatto con l’esterno. E' possibile utilizzare l'uscita di un sistema GC o HPLC come ingresso dello spettrometro di massa.
 è necessario per impedire una perdita di ionizzazione per urto con i gas atmosferici. Introduzione del campione. L’introduzione del campione nella camera di ionizzazione può essere fatta sia allo stato solido, usando una sonda, che allo stato liquido o gassoso, usando un sistema di valvole che permettono di accedere alla camera di ionizzazione senza che questa venga a contatto con l’esterno. E possibile utilizzare l uscita di un sistema GC o HPLC come ingresso dello spettrometro di massa.")
59
Camera di ionizzazione
Se una molecola è investita in fase vapore da un fascio di elettroni di notevole energia cinetica si può avere per urto la sua ionizzazione a ione positivo o negativo. In genere gli strumenti sono regolati per lavorare unicamente con ioni positivi, i quali possono spontaneamente o per urto decomporsi in una serie di frammenti di massa inferiore e questi a loro volta in altri. Ogni molecola avrà quindi una sua frammentazione caratteristica e specifica che dipenderà sia dalla natura delle molecole sia dalle condizioni operative di ionizzazione. Il campione viene ionizzato in un’apposita camera di ionizzazione, in cui il fascio di elettroni viene prodotto da una sorgente ionica che varia a seconda della tecnica utilizzata. In genere gli elettroni sono emessi da un filamento caldo di tungsteno o renio, e passano attraverso un condotto, che crea il raggio, nella parte centrale della camera che contiene il campione gassoso. La frazione di elettroni che non urta contro le molecole è raccolta da una trappola per gli elettroni, le molecole che non sono ionizzate sono allontanate dalla pompa ad alto vuoto, mentre quelle ionizzate sono accelerate e convogliate verso l’analizzatore.

60
Tecniche di ionizzazione
IMPATTO ELETTRONICO (E.I.) La ionizzazione per impatto elettronico è la tecnica più comune. Un filamento di tungsteno incandescente emette un fascio di elettroni che, accelerati verso un anodo posto dalla parte opposta al filamento, acquistano un’elevata energia (ca. 70 eV). Quando questi elettroni vengono a contatto con la sfera elettronica di una molecola (impatto elettronico), le trasferiscono la loro energia, provocando l’espulsione di un elettrone con formazione di un radical catione (ione molecolare) M+•. Siccome l’energia necessaria per ionizzare una molecola organica è di circa eV, i radical cationi sono prodotti ad un’energia vibrazionale molto alta, che ne può determinare la frammentazione con formazione di un radicale e un catione. Tutti gli ioni positivi (cationi e radical cationi) sono respinti da una piastra, tenuta ad un potenziale positivo, verso una serie di piastre forate, tenute a potenziale positivo crescente, dette piastre acceleratrici. Nel loro tragitto gli ioni subiscono un’accelerazione proporzionale al potenziale V delle piastre acceleratrici e vengono espulsi, attraverso una fenditura di uscita. e- M0+ . Radical catione ione molecolare M R M1+ Radicale + catione (frammento)
 La ionizzazione per impatto elettronico è la tecnica più comune. Un filamento di tungsteno incandescente emette un fascio di elettroni che, accelerati verso un anodo posto dalla parte opposta al filamento, acquistano un’elevata energia (ca. 70 eV). Quando questi elettroni vengono a contatto con la sfera elettronica di una molecola (impatto elettronico), le trasferiscono la loro energia, provocando l’espulsione di un elettrone con formazione di un radical catione (ione molecolare) M+•. Siccome l’energia necessaria per ionizzare una molecola organica è di circa eV, i radical cationi sono prodotti ad un’energia vibrazionale molto alta, che ne può determinare la frammentazione con formazione di un radicale e un catione. Tutti gli ioni positivi (cationi e radical cationi) sono respinti da una piastra, tenuta ad un potenziale positivo, verso una serie di piastre forate, tenute a potenziale positivo crescente, dette piastre acceleratrici. Nel loro tragitto gli ioni subiscono un’accelerazione proporzionale al potenziale V delle piastre acceleratrici e vengono espulsi, attraverso una fenditura di uscita. e- M0+ . Radical catione ione molecolare. M. R. M1+ Radicale + catione (frammento)")
61
Tecniche di ionizzazione
IONIZZAZIONE CHIMICA (C.I.) La ionizzazione chimica viene utilizzata quando gli ioni molecolari prodotti con il metodo dell’impatto elettronico sono troppo poco stabili e si frammentano completamente. Si basa sull’interazione del campione vaporizzato con un reagente ionizzato. I piu’ usati sono quelli che derivano dalla ionizzazione ad impatto elettronico del metano. Se la molecola M ha un’affinità per il protone più alta di quella del metano, allora si avrà la formazione dello ione M-H+. Gli ioni M-H (detti quasimolecolari) non possiedono una energia così elevata e quindi subiscono una minore frammentazione. IONIZZAZIONE ELETTROSPRAY (E.S.I.) Il campione, sciolto in un solvente polare, è nebulizzato a pressione atmosferica dentro la camera di ionizzazione attraverso un ago tenuto ad un alto potenziale elettrico. Le goccioline di spray, che si sono caricate positivamente per azione del campo elettrico, vengono attratte verso una "lente di estrazione di ioni", che grossolanamente è costituito da un capillare mantenuto sotto vuoto e a un potenziale negativo; in tal modo il sovente evapora e gli ioni carichi sono accelerati verso l'analizzatore. Questa tecnica di ionizzazione è largamente usata negli strumenti HPLC-MS.
 La ionizzazione chimica viene utilizzata quando gli ioni molecolari prodotti con il metodo dell’impatto elettronico sono troppo poco stabili e si frammentano completamente. Si basa sull’interazione del campione vaporizzato con un reagente ionizzato. I piu’ usati sono quelli che derivano dalla ionizzazione ad impatto elettronico del metano. Se la molecola M ha un’affinità per il protone più alta di quella del metano, allora si avrà la formazione dello ione M-H+. Gli ioni M-H (detti quasimolecolari) non possiedono una energia così elevata e quindi subiscono una minore frammentazione. IONIZZAZIONE ELETTROSPRAY (E.S.I.) Il campione, sciolto in un solvente polare, è nebulizzato a pressione atmosferica dentro la camera di ionizzazione attraverso un ago tenuto ad un alto potenziale elettrico. Le goccioline di spray, che si sono caricate positivamente per azione del campo elettrico, vengono attratte verso una lente di estrazione di ioni , che grossolanamente è costituito da un capillare mantenuto sotto vuoto e a un potenziale negativo; in tal modo il sovente evapora e gli ioni carichi sono accelerati verso l analizzatore. Questa tecnica di ionizzazione è largamente usata negli strumenti HPLC-MS.")
62
Analizzatori per MS ANALIZZATORE MAGNETICO
E' l'analizzatore più usato, perchè consente di ottenere le risoluzioni migliori. E’ costituito da un tubo lungo circa 1 metro, piegato con un raggio di curvatura r' ed immerso in un campo magnetico H. Gli ioni che escono dalla camera di ionizzazione entrano nel tubo analizzatore e, per effetto del campo magnetico, subiscono una deviazione dalla loro traiettoria rettilinea (deflessione). La nuova traiettoria curvilinea ha un raggio di curvatura r che è direttamente proporzionale alla quantità di moto dello ione (mv) e inversamente proporzionale al campo magnetico H. Di conseguenza per un certo valore della coppia H e V esisterà un solo valore di massa m per cui il raggio di deflessione r coincide con il raggio di curvatura del tubo r'. Gli ioni che hanno questo valore di massa escono dal tubo, gli altri no. Operando a potenziale V costante e facendo una scansione di campo H è possibile fare uscire dal tubo gli ioni a diversa massa in tempi diversi. Gli ioni che escono dal tubo vengono raccolti da un fotomoltiplicatore, che traduce l’intensità degli ioni in corrente elettrica (Rivelatore). Gli strumenti sono tarati (si usano dei perfluorocheroseni) in modo che a ciascun valore di campo corrisponda un certo valore di massa. Si ottiene così lo spettro di massa, che è un istogramma che riporta in ascisse i valori di massa crescente (gli strumenti sono tarati in genere da 30 a 1000 uma) e in ordinate la corrente ionica. ANALIZZATORE A QUADRUPOLO E’ costituito da quattro barre cilindriche metalliche, lunghe circa 20 cm., che delimitano il "cammino" degli ioni provenienti dalla camera di ionizzazione e diretti al detector. Le barre sono mantenute ad un potenziale elettromagnetico oscillante, in modo che quando le due sbarre verticali hanno potenziale positivo quelle orizzontali l’hanno negativo, e viceversa. Gli elettroni, accelerati dalle piastre acceleratrici, entrano nel tunnel delimitato dalle barre e vengono respinti dai poli positivi ed attratti dai negativi. Tuttavia, a causa dell’oscillazione del quadrupolo gli ioni assumono una traiettoria a zig zag e finiscono con lo scaricarsi su una delle barre, tranne quelli che, per una certo valore di frequenza di oscillazione, hanno un’energia cinetica tale per cui il moto diventa sinusoidale e riescono ad uscire dal tunnel ed entrare nel sistema di rivelazione (fotomoltiplicatore). Operando quindi una scansione di frequenza di oscillazione del campo è possibile far uscire ioni a massa molecolare crescente.
. La nuova traiettoria curvilinea ha un raggio di curvatura r che è direttamente proporzionale alla quantità di moto dello ione (mv) e inversamente proporzionale al campo magnetico H. Di conseguenza per un certo valore della coppia H e V esisterà un solo valore di massa m per cui il raggio di deflessione r coincide con il raggio di curvatura del tubo r . Gli ioni che hanno questo valore di massa escono dal tubo, gli altri no. Operando a potenziale V costante e facendo una scansione di campo H è possibile fare uscire dal tubo gli ioni a diversa massa in tempi diversi. Gli ioni che escono dal tubo vengono raccolti da un fotomoltiplicatore, che traduce l’intensità degli ioni in corrente elettrica (Rivelatore). Gli strumenti sono tarati (si usano dei perfluorocheroseni) in modo che a ciascun valore di campo corrisponda un certo valore di massa. Si ottiene così lo spettro di massa, che è un istogramma che riporta in ascisse i valori di massa crescente (gli strumenti sono tarati in genere da 30 a 1000 uma) e in ordinate la corrente ionica. ANALIZZATORE A QUADRUPOLO. E’ costituito da quattro barre cilindriche metalliche, lunghe circa 20 cm., che delimitano il cammino degli ioni provenienti dalla camera di ionizzazione e diretti al detector. Le barre sono mantenute ad un potenziale elettromagnetico oscillante, in modo che quando le due sbarre verticali hanno potenziale positivo quelle orizzontali l’hanno negativo, e viceversa. Gli elettroni, accelerati dalle piastre acceleratrici, entrano nel tunnel delimitato dalle barre e vengono respinti dai poli positivi ed attratti dai negativi. Tuttavia, a causa dell’oscillazione del quadrupolo gli ioni assumono una traiettoria a zig zag e finiscono con lo scaricarsi su una delle barre, tranne quelli che, per una certo valore di frequenza di oscillazione, hanno un’energia cinetica tale per cui il moto diventa sinusoidale e riescono ad uscire dal tunnel ed entrare nel sistema di rivelazione (fotomoltiplicatore). Operando quindi una scansione di frequenza di oscillazione del campo è possibile far uscire ioni a massa molecolare crescente.")
63
Lo spettro di massa si presenta quindi come un insieme di linee verticali (picchi) di intensità diversa, ciascuna corrispondente al valore di massa di uno ione frammento. Il picco a valore di massa più elevato è quello relativo allo ione molecolare. Dallo spettro di massa si può risalire dunque alla struttura di un composto incognito, attribuendo ai singoli ioni una composizione elementare e ricostruendo i meccanismi di frammentazione seguendo schemi tipici per le varie classi di composti. Innanzitutto si identifica lo ione molecolare. Tuttavia il picco può essere poco intenso o addirittura assente nel caso di molecole facilmente frammentabili (l’intensità del picco dipende dalla stabilità della specie che lo genera); la sua intensità è maggiore per molecole lineari e minore per molecole ramificate, inoltre in una serie omologa diminuisce all'aumentare della massa molecolare. Il picco dello ione molecolare è spesso accompagnato da altri picchi, in genere più deboli, a massa M + 1, M + 2, ecc. dovuti alle molecole contenenti isotopi degli elementi che le costituiscono. La maggior parte degli elementi che compongono i composti organici, infatti, possiede diversi isotopi naturali, di cui di solito il più leggero è il più abbondante. Gli altri picchi corrispondono a ioni-frammento derivati per frammentazione dello ione molecolare. L’altezza dei picchi è normalizzata a 100. Il picco alto 100 è il picco base; corrisponde allo ione-frammento più stabile, che può essere o meno lo ione molecolare. Nella camera di ionizzazione può succedere che alcuni ioni subiscono un ulteriore strappo di elettroni, con formazione di ioni doppiamente carichi (M++). Questi vengono focalizzati come se fossero ioni di massa (M/2)+. Nonostante che questo fenomeno sia molto raro, è comunque corretto mettere in ascisse non la massa m, ma il rapporto m/z, dove z è la carica dello ione
; la sua intensità è maggiore per molecole lineari e minore per molecole ramificate, inoltre in una serie omologa diminuisce all aumentare della massa molecolare. Il picco dello ione molecolare è spesso accompagnato da altri picchi, in genere più deboli, a massa M + 1, M + 2, ecc. dovuti alle molecole contenenti isotopi degli elementi che le costituiscono. La maggior parte degli elementi che compongono i composti organici, infatti, possiede diversi isotopi naturali, di cui di solito il più leggero è il più abbondante. Gli altri picchi corrispondono a ioni-frammento derivati per frammentazione dello ione molecolare. L’altezza dei picchi è normalizzata a 100. Il picco alto 100 è il picco base; corrisponde allo ione-frammento più stabile, che può essere o meno lo ione molecolare. Nella camera di ionizzazione può succedere che alcuni ioni subiscono un ulteriore strappo di elettroni, con formazione di ioni doppiamente carichi (M++). Questi vengono focalizzati come se fossero ioni di massa (M/2)+. Nonostante che questo fenomeno sia molto raro, è comunque corretto mettere in ascisse non la massa m, ma il rapporto m/z, dove z è la carica dello ione.")
64
Cromatogramma Il segnale in uscita dal rivelatore passa ad un registratore che ha il compito di realizzare il tracciato cromatografico. Ogni sostanza in uscita dalla colonna genera un segnale che verrà registrato sotto forma di 'picco‘ caratterizzato da un’altezza (distanza fra il massimo del picco e la sua base, misurata perpendicolarmente all’asse dei tempi) e da un’ampiezza (segmento delimitato sulla base del picco dai punti di intersezione delle tangenti tracciate nei punti di flesso di ambedue i lati). La successione dei vari picchi, corrispondenti alle varie sostanze in uscita dalla colonna, costituisce il 'cromatogramma‘, dove in ordinate é riportata la risposta del rivelatore e in ascisse i tempi di uscita delle varie sostanze. Oltre ad altezza ed ampiezza, si determinano due parametri essenziali: tempo di ritenzione: tempo impiegato tra l’iniezione del campione e la registrazione del massimo del picco; area del picco: superficie delimitata dal contorno del picco e la linea di base;
 e da un’ampiezza (segmento delimitato sulla base del picco dai punti di intersezione delle tangenti tracciate nei punti di flesso di ambedue i lati). La successione dei vari picchi, corrispondenti alle varie sostanze in uscita dalla colonna, costituisce il. cromatogramma‘, dove in ordinate é riportata la risposta del rivelatore e in ascisse i tempi di uscita delle varie sostanze. Oltre ad altezza ed ampiezza, si determinano due parametri essenziali: tempo di ritenzione: tempo impiegato tra l’iniezione del campione e la registrazione del massimo del picco; area del picco: superficie delimitata dal contorno del picco e la linea di base;")
65
Proprietà colonna e cromatogramma
La selettività è definita come la capacità di una colonna di fornire picchi distanziati e dipende dalla temperatura e dalla natura della fase stazionaria. L’ efficienza è la capacità del sistema cromatografico di mantenere compatta la banda di eluizione di una sostanza lungo tutto il percorso della fase mobile. Ciò significa ottenere picchi alti e stretti all’uscita della colonna. La cosa é di grande importanza, perché qualora due sostanze avessero tempi di ritenzione molto vicini se ne potrebbe ottenere ugualmente la separazione. La risoluzione tiene conto sia della selettività che dell’efficienza, e indica il grado di effettiva separazione ottenuto per due sostanze in un processo cromatografico. Dal punto di vista numerico si ottiene dalla relazione: Per avere una buona separazione, dal punto di vista quantitativo, si deve avere risoluzione almeno 0,8 .

66
Esempi di diversa risoluzione
Per l’analisi quantitativa è preferibile una risoluzione uguale o superiore a 1.5

67
Esempi di diverse risoluzioni

68
Gascromatografia: analisi qualitativa / quantitativa
I metodi utilizzabili per l’individuazione delle sostanze sono: • Basarsi su dati di letteratura, quali i tempi di ritenzione; purtroppo tali valori dipendono da molti fattori quali le caratteristiche dello strumento, le condizioni operative e l’operatore. • Effettuare un confronto dei tempi di ritenzione tra la miscela in esame e sostanze pure o miscele di composizione nota. • Metodo basato sull’arricchimento. Quando si ritiene che un determinato picco corrisponda ad una sostanza nota, si aggiunge alla miscela una certa quantità di sostanza pura. Se compare un altro picco, siamo sicuri che la specie nota non é presente nella miscela, mentre se un picco risulta più alto, potrebbe essere presente e per questo é necessario effettuare altre analisi cambiando condizioni operative. Analisi quantitativa L'analisi quantitativa ha l’obiettivo di valutare le concentrazioni Utilizza come dato l’area dei picchi . Si usano due metodi: - STANDARD INTERNO - STANDARD ESTERNO

69
Metodo dello standard interno
Questo metodo consente di ottenere risultati molto accurati e non affetti da errore legato alla quantità iniettata in colonna, in quanto sfrutta il rapporto tra l'area del picco dell'analita e l'area del picco di una sostanza (“standard interno”) appositamente aggiunto in quantità nota. Si prepara una soluzione standard utilizzando due composti, dei quali uno deve essere il componente che interessa analizzare, l’altro é invece un composto, detto standard interno, che deve obbedire ai seguenti requisiti: • non essere presente nella miscela da analizzare; • essere ben risolto dagli altri componenti; • avere un tempo di ritenzione simile a quello della sostanza da determinare; • non reagire con nessun componente della miscela. Si preparano più soluzioni note con diversi rapporti tra analita e standard interno e si costruisce una retta di lavoro avente come ordinata il rapporto tra le aree dell’analita e dello standard interno.
 appositamente aggiunto in quantità nota. Si prepara una soluzione standard utilizzando due composti, dei quali uno deve essere il componente che. interessa analizzare, l’altro é invece un composto, detto standard interno, che deve obbedire ai seguenti requisiti: • non essere presente nella miscela da analizzare; • essere ben risolto dagli altri componenti; • avere un tempo di ritenzione simile a quello della sostanza da determinare; • non reagire con nessun componente della miscela. Si preparano più soluzioni note con diversi rapporti tra analita e standard interno e si costruisce una retta di lavoro avente come ordinata il rapporto tra le aree dell’analita e dello standard interno.")
70
Metodo dello standard interno
Caso del benzene Il metodo si basa sulla determinazione del fattore di risposta relativo RRF del benzene rispetto al benzene perdeuterato (standard interno aggiunto al campione prima del desorbimento termico) eseguita per diverse quantità di analita: RRFi = 2.5 * Bi/D Ove: RRFi = fattore di risposta RRF calcolato per la quantità i-esima di benzene (in ng); Bi = Area del picco cromatografico relativo al benzene per la quantità i-esima, in unità arbitrarie; D = Area del picco cromatografico relativo al benzene perdeuterato (quantità fissa di 2.5ng). Per la calibrazione del rivelatore si fa ricorso ad un "riferimento primario", costituito da un contenitore ermetico contenente benzene in concentrazione certificata. Per aliquote differenti di "riferimento primario" (tutte contenenti la stessa concentrazione di standard interno) si registrano le risposte strumentali, espresse in termini di "area di picco cromatografico“. Da queste ultime si calcolano i RRFi. Graficando i valori degli RRFi verso le corrispondenti quantità di benzene rivelate, si costruisce la "curva di taratura" e si definisce il campo di linearità strumentale, entro il quale occorre eseguire le determinazioni quantitative del benzene nell'aria campione. Si sottopone ad analisi una prima aliquota di aria campione, e si ricava il valore di RRFx, ovvero il fattore di risposta del benzene nel campione rispetto allo standard interno. Riportando il valore di RRFx nella curva di calibrazione, si ricava la quantità di benzene incognita Qx.
 eseguita per diverse quantità di analita: RRFi = 2.5 * Bi/D. Ove: RRFi = fattore di risposta RRF calcolato per la quantità i-esima di benzene (in ng); Bi = Area del picco cromatografico relativo al benzene per la quantità i-esima, in unità arbitrarie; D = Area del picco cromatografico relativo al benzene perdeuterato (quantità fissa di 2.5ng). Per la calibrazione del rivelatore si fa ricorso ad un riferimento primario , costituito da un contenitore ermetico contenente benzene in concentrazione certificata. Per aliquote differenti di riferimento primario (tutte contenenti la stessa concentrazione di standard interno) si registrano le risposte strumentali, espresse in termini di area di picco cromatografico . Da queste ultime si calcolano i RRFi. Graficando i valori degli RRFi verso le corrispondenti quantità di benzene rivelate, si costruisce la curva di taratura e si definisce il campo di linearità strumentale, entro il quale occorre eseguire le determinazioni quantitative del benzene nell aria campione. Si sottopone ad analisi una prima aliquota di aria campione, e si ricava il valore di RRFx, ovvero il fattore di risposta del benzene nel campione rispetto allo standard interno. Riportando il valore di RRFx nella curva di calibrazione, si ricava la quantità di benzene incognita Qx.")
71
Ottimizzazione di una analisi di GC
Scelta della colonna. Per campioni con sostanze di polarità analoga ma con punti di ebollizione abbastanza diversi, non è necessario che la fase stazionaria sia particolarmente selettiva, per cui se ne impiega una apolare. In questo modo i composti usciranno in base alla loro volatilità decrescente. Per campioni con sostanze di polarità diversa ma con punti di ebollizione abbastanza vicini, si possono usare fasi stazionarie sia polari che apolari. Infatti, con fasi polari sarà il componente polare a venir maggiormente trattenuto e l’altro uscirà per primo, mentre con fase apolare avverrà l’inverso. Per campioni contenenti contemporaneamente sostanze non polari e sostanze non polari ma polarizzabili (ad esempio una miscela esano-benzene), si utilizzano fasi molto polari. Quest’ultime infatti, polarizzano i composti aromatici (come il benzene) stabilendo legami dipolo-dipolo indotto, mentre non trattengono, ad esempio, l’esano che é assolutamente apolare e non polarizzabile, per cui sarà l’esano ad uscire per primo. Scelta della temperatura della colonna. Vi sono due possibilità di impostare la temperatura. Si può fare una isoterma, cioè la temperatura rimane sempre la stessa, oppure possiamo impostare una 'programmata', in cui la temperatura é variabile. La programmazione si esegue fissando la Temperatura iniziale Ti, il Tempo di permanenza alla temperatura iniziale, la Temperatura massima finale da raggiungere Tf, la Velocità di incremento della temperatura (gradi/min) ed il Tempo di permanenza alla temperatura finale Scelta della temperatura della camera di iniezione. Questa deve essere in grado di vaporizzare l’intera miscela di composti quindi deve essere superiore alla temperatura di ebollizione del composto più altobollente.
, si utilizzano fasi molto polari. Quest’ultime infatti, polarizzano i composti aromatici (come il benzene) stabilendo legami dipolo-dipolo indotto, mentre non trattengono, ad esempio, l’esano che é assolutamente apolare e non polarizzabile, per cui sarà l’esano ad uscire per primo. Scelta della temperatura della colonna. Vi sono due possibilità di impostare la temperatura. Si può fare una isoterma, cioè la temperatura rimane. sempre la stessa, oppure possiamo impostare una programmata , in cui la temperatura é variabile. La. programmazione si esegue fissando la Temperatura iniziale Ti, il Tempo di permanenza alla temperatura iniziale, la Temperatura massima finale da raggiungere Tf, la Velocità di incremento della temperatura (gradi/min) ed il Tempo di permanenza alla temperatura finale. Scelta della temperatura della camera di iniezione. Questa deve essere in grado di vaporizzare l’intera miscela di composti quindi deve essere superiore alla temperatura di ebollizione del composto più altobollente.")
72
Ottimizzazione di una analisi di GC
Scelta del carrier e della sua portata. La scelta del gas di trasporto dipende in misura preponderante dal tipo di rivelatore utilizzato, ma va tenuto presente che l’efficienza aumenta utilizzando gas con peso molecolare elevato. La portata del gas eluente (carrier) ha influenza sull’efficienza della colonna, sui tempi di ritenzione e sulla risposta del rivelatore. In linea di massima il tempo di ritenzione é inversamente proporzionale alla velocità media del gas di trasporto. Il rivelatore produce segnali inversamente proporzionali al volume di gas che passa, quindi l’area del picco diminuisce con l’aumentare della portata. I flussi ottimali si aggirano intorno a mL/min. Trattamento del campione. Spesso il campione da esaminare non può essere iniettato come tale o in soluzione. Alcuni accorgimenti sono i seguenti: a) Disidratazione. Quando si opera con fasi stazionarie particolarmente sensibili all’umidità e si usano rivelatori (ECD) che ne risentono negativamente, é necessario procedere alla disidratazione dei campioni. b) Derivatizzazione. L’analisi di composti altobollenti richiede temperature troppo elevate con rischi di decomposizione, polimerizzazioni o addirittura carbonizzazioni. In questi casi può essere molto utile la modificazione chimica del campione che permette di ottenere derivati a maggior volatilità. c) Cromatografia dello spazio di testa. Quando si devono analizzare tracce di composti volatili in campioni solidi o in una grande massa di solvente, il miglior modo é quello di iniettare il vapore che si trova in equilibrio con il campione da analizzare.
 ha influenza sull’efficienza della colonna, sui tempi di ritenzione e sulla risposta del rivelatore. In linea di massima il tempo di ritenzione é inversamente proporzionale alla velocità media del gas di trasporto. Il rivelatore produce segnali inversamente proporzionali al volume di gas che passa, quindi l’area del picco diminuisce con l’aumentare della portata. I flussi ottimali si aggirano intorno a mL/min. Trattamento del campione. Spesso il campione da esaminare non può essere iniettato come tale o in soluzione. Alcuni accorgimenti sono i seguenti: a) Disidratazione. Quando si opera con fasi stazionarie particolarmente sensibili all’umidità e si usano rivelatori (ECD) che ne risentono negativamente, é necessario procedere alla disidratazione dei campioni. b) Derivatizzazione. L’analisi di composti altobollenti richiede temperature troppo elevate con rischi di decomposizione, polimerizzazioni o addirittura carbonizzazioni. In questi casi può essere molto utile la modificazione chimica del campione che permette di ottenere derivati a maggior volatilità. c) Cromatografia dello spazio di testa. Quando si devono analizzare tracce di composti volatili in campioni. solidi o in una grande massa di solvente, il miglior modo é quello di iniettare il vapore che si trova in equilibrio con il campione da analizzare.")
73
Metodo GC per Benzene La separazione del benzene è realizzata per gascromatografia in colonna capillare. Quale gas di trasporto si usa elio iperpuro. Numerose colonne gascromatografiche sono in grado di separare efficacemente il benzene dagli altri composti organici volatili presenti nell'atmosfera urbana. Esse sono raggruppabili in due tipi: a) colonne capillari del tipo "Plot", la cui fase stazionaria è costituita da un film di allumina porosa drogata con sali (Al203/KCl, Al203/Na2S04); b) colonne capillari ricoperte di siliconi polimeri chimicamente legati (tipo DB1, DB5, CPSil5, CPSil8, SP1, HPl, HPS, etc.). Per la separazione del benzene dagli altri composti organici volatili si usa di una colonna capillare tipo DBl (gomma di metilpolisilossano chimicamente legato) avente lunghezza di 60m diametro interno 0.32mm e spessore di film 0.33um. Eluizione gascromatografica. L'eluizione del benzene nella colonna separativa è eseguita in gradiente di temperatura secondo il seguente programma: a) temperatura iniziale 5°C, isoterma iniziale 3min.; b) primo incremento termico +3°C/min. fino a 50°C; c) secondo incremento termico +5°C/min. fino a 210°C; Come detector si usa il principio della spettometria di massa operante in rivelazione selettiva di ioni ("Selected Ion Monitoring - Mass Spectrometry" o SIM-MS).
 colonne capillari del tipo Plot , la cui fase stazionaria è costituita da un film di allumina porosa drogata con sali (Al203/KCl, Al203/Na2S04); b) colonne capillari ricoperte di siliconi polimeri chimicamente legati (tipo DB1, DB5, CPSil5, CPSil8, SP1, HPl, HPS, etc.). Per la separazione del benzene dagli altri composti organici volatili si usa di una colonna capillare tipo DBl (gomma di metilpolisilossano chimicamente legato) avente lunghezza di 60m diametro interno 0.32mm e spessore di film 0.33um. Eluizione gascromatografica. L eluizione del benzene nella colonna separativa è eseguita in gradiente di temperatura secondo il seguente programma: a) temperatura iniziale 5°C, isoterma iniziale 3min.; b) primo incremento termico +3°C/min. fino a 50°C; c) secondo incremento termico +5°C/min. fino a 210°C; Come detector si usa il principio della spettometria di massa operante in rivelazione selettiva di ioni ( Selected Ion Monitoring - Mass Spectrometry o SIM-MS).")
74
fotometria ultravioletta
OZONO – O3 Metodo d’analisi UNI EN 14625:2005 Qualità dell'aria ambiente - Metodo normalizzato per la misurazione della concentrazione di ozono mediante fotometria ultravioletta Principio del metodo: Un raggio UV di lunghezza d'onda di 254 nm UV viene inviato attraverso la cella di campionatura dove è assorbito in proporzione alla quantità di Ozono presente.

75
Il metodo ad assorbimento UV si basa sulla proprietà dell’Ozono di assorbire raggi ultravioletti nell’area dei 254 nm (Legge di Lambert-Beer). La misura è ottenuta tramite continue ed alternate iniezioni di gas di riferimento e di gas da analizzare nella cella di analisi mediante una valvola a solenoide di lunga durata. Tale metodo viene chiamato “cross flow modulation”. Il sistema di purificazione dell’aria di riferimento è immune alle interferenze dell’umidità.

76
PARTICOLATO Metodo gravimetrico PM10: frazione grossolana
PM2,5: frazione fine Il particolato totale e le frazioni PM10 e PM2,5 vengono misurati mediante raccolta su filtro per 24 ore in condizioni standardizzate e successiva determinazione gravimetrica delle polveri filtrate. Nel caso della frazione PM10 e PM2,5 la testa della apparecchiatura di prelievo ha una particolare geometria definita in modo tale che sul filtro arrivino e siano trattenute le particelle secondo una curva di efficienza di raccolta definita Il metodo gravimetrico è non distruttivo e per questo consente la successiva analisi chimico-fisica del particolato per la speciazione chimica.

77
Schema di un campionatore di PM
I campionatori gravimetrici di PM sono costituiti da: • una testa selettiva, in grado di selezionare le particelle con diametro inferiore a 10 μm (PM10) o 2.5 μm (PM2.5) • un sistema di controllo del flusso d’aria capace di mantenere una velocità costante nell’aspiratore (campionamento isocinetico), • un porta filtri con filtro di raccolta
 o 2.5 μm (PM2.5) • un sistema di controllo del flusso d’aria capace di mantenere una velocità costante nell’aspiratore (campionamento isocinetico), • un porta filtri con filtro di raccolta.")
78
Testa di prelievo Il materiale particellare PM10 viene selezionato dalla testa di prelievo attraverso nove ugelli (ugelli di accelerazione particellare), in cui passa l’aria aspirata dal campionatore, strutturati in modo da provocare variazioni nella velocità delle particelle. In questo modo, quando le particelle passano nella zona di frazionamento sottostante, quelle con diametro superiore a 10 μm si depositano sulla base metallica (ricoperta con un sottile strato di silicone), mentre quelle con diametro inferiore rimangono in sospensione nel flusso d’aria e condotte attraverso un'altra serie di ugelli nell’impattore a cascata posizionato nell’alloggiamento del portafiltro. In una testa di prelievo per PM-10 l’efficienza di campionamento per le particelle inferiori a 10µm è di gran lunga più elevata rispetto a quella delle particelle di diametro maggiore.
, in cui passa l’aria aspirata dal campionatore, strutturati in modo da provocare variazioni nella velocità delle particelle. In questo modo, quando le particelle passano nella zona di frazionamento sottostante, quelle con diametro superiore a 10 μm si depositano sulla base metallica (ricoperta con un sottile strato di silicone), mentre quelle con. diametro inferiore rimangono in sospensione nel flusso d’aria e condotte attraverso un altra serie di ugelli nell’impattore a cascata posizionato nell’alloggiamento del portafiltro. In una testa di prelievo per PM-10 l’efficienza di campionamento per le particelle inferiori a 10µm è di gran lunga più elevata rispetto a quella delle particelle di diametro maggiore.")
79
Misura per pesata PM Il filtro viene pesato prima e dopo il campionamento e la quantità che ne deriva viene rapportata al volume di aria campionata e normalizzata a 25 °C ed 1 atm. Il locale di pesatura e condizionamento dovrebbe essere preferibilmente lo stesso • forno di essiccazione termicamente controllato (stabilità±5.0 °C) • bilancia analitica con risoluzione minima di 0.01 mg (per PM10 almeno1 μg) • essiccatori in vetro o ceramica con gel di silice Procedura: 1. Condizionamento in forno per almeno 3 ore a 160°C 2. Essiccamento in gel di silice per almeno 12 ore 3. Pesatura 4. Campionamento 5. Ripetere i punti
 • bilancia analitica con risoluzione minima di 0.01 mg (per PM10 almeno1 μg) • essiccatori in vetro o ceramica con gel di silice. Procedura: 1. Condizionamento in forno per almeno 3 ore a 160°C. 2. Essiccamento in gel di silice per almeno 12 ore. 3. Pesatura. 4. Campionamento. 5. Ripetere i punti")
80
Qualità delle misure gravimetriche PM
Il limite inferiore della concentrazione di massa da misurare è determinato dalla ripetibilità dei pesi del filtro vuoto. Il limite superiore è invece corrispondente al valore di massa di particolato al di sopra della quale il campionatore non riesce a mantenere la portata di campionamento all’interno dei limiti permessi a causa dell’elevata caduta di pressione sul filtro. Pertanto non di immediata definizione, ma dipende dalla distribuzione dimensionale delle particelle, dall’umidità, dal tipo di filtro, etc… Fonti di errore: - Perdita delle particelle volatili durante le operazioni di conservazione dei filtri. - Ritenzione sul filtro di specie gassose, quali biossido di zolfo e acido nitrico. La ritenzione di SO2, seguita dall’ossidazione a solfati, è limitata utilizzando filtri poco alcalini. La ritenzione di acido nitrico è stata osservata su diversi mezzi filtranti (fibra di vetro, fibra di quarzo, estere di cellulosa).
.")
81
ANALIZZATORI AUTOMATICI PM
Essi si basano su principi d’inerzia, ottici o d’attenuazione beta. Ne esistono diversi: •lo smoke shade e’un campionatore automatico, che determina il particolato attraverso l’assorbimento della luce della macchia costituita dalle particelle raccolte sul filtro. •il campionatore a nastro e’un campionatore ottico, automatico, più sensibile rispetto al precedente, che si basa sulla trasmissione della luce attraverso il particolato che si e’depositato. •l’analizzatore a microbilancia, il cosiddetto TEOM •gli analizzatori ad attenuazione Beta, che sono campionatori che si basano sul principio dell’attenuazione dell’energia posseduta da un fascio di elettroni, quando essi attraversano uno strato sottile di materiale. Se ne conoscono diversi, tra cui l’Adam, l’Andersen il Wedding •gli analizzatori che si basano sul principio della bilancia oscillante, come il Nefelometro scattering della luce

82
Analizzatori beta Gli analizzatori di PM10 automatici cosiddetti "beta" determinano la massa di particolato mediante la misurazione dell'attenuazione di basse radiazioni ß prodotte da una sorgente radioattiva interna allo strumento. I raggi beta emessi da una sorgente radioattiva di carbonio 14 attraversano il filtro "bianco" e alla fine del ciclo di 24 ore il filtro campionato, la differenza nell'assorbimento dei raggi beta da parte del filtro è proporzionale alla concentrazione del PM10 campionato. Gli analizzatori automatici sono costituiti da un nastro di prelievo che può essere del tipo a carta continua o a catena di supporti metallici di porta filtri. L'aria da analizzare viene aspirata attraverso il sistema filtrante in modo da trattenere le polveri sul nastro filtrante. All'inizio di ogni ciclo e al termine dello stesso il rivelatore determinerà l'assorbimento dei raggi beta emessi dalla sorgente da parte della polvere depositata sul filtro, essendo questa proporzionale al quantitativo di polvere presente ed in funzione del volume di aria filtrata l'analizzatore rilascerà il valore di concentrazione.

83
Analisi metalli pesanti SPETTROSCOPIA DI ASSORBIMENTO ATOMICO
FA/AAS: con atomizzazione di fiamma (generalmente ottenuta con acetilene ed aria) HGA/AAS: con atomizzazione elettrotermica (ottenuta mediante una corrente elettrica ad alta potenza che crea un riscaldamento per effetto Joule).
 HGA/AAS: con atomizzazione elettrotermica (ottenuta mediante una corrente elettrica ad alta potenza che crea un riscaldamento per effetto Joule).")
84
SPETTROSCOPIA DI ASSORBIMENTO ATOMICO
Questa tecnica è basata sull’esame dell’assorbimento di una radiazione elettromagnetica dopo che questa passa in un mezzo in cui il campione sia presente come atomi o ioni monoatomici. Quando un atomo viene posto nelle condizioni di acquistare energia elettromagnetica di intensità adeguata, uno o più elettroni esterni posso infatti abbandonare gli orbitali in cui abitualmente si trovano, per venire promossi ad orbitali più ricchi di energia. Di conseguenza l’atomo, che si trovava nella sua configurazione elettronica normale (o stato energetico fondamentale) raggiunge un livello energetico più ricco di energia e quindi meno stabile (stato eccitato). Da questo stato eccitato l’atomo decade rapidamente, tornando allo stato fondamentale e restituendo all’ambiente l’energia appena acquistata (prevalentemente per via termica, nel senso che l’energia accumulata viene dispersa mediante urti con particelle che si trovano nelle vicinanze degli elettroni eccitati). Ogni atomo disponendo di un proprio numero di elettroni situati su determinati orbitali, è caratterizzato da specifici spettri di assorbimento atomico, costituiti da una serie di righe di risonanza, la cui intensità è proporzionale alla probabilità della transizione elettronica corrispondente. Ogni transizione, infatti, corrisponde all’assorbimento di una precisa energia e produce una riga spettrale con una caratteristica lunghezza d’onda.
 raggiunge un livello energetico più ricco di energia e quindi meno stabile (stato eccitato). Da questo stato eccitato l’atomo decade rapidamente, tornando allo stato fondamentale e restituendo all’ambiente l’energia appena acquistata (prevalentemente per via termica, nel senso che l’energia accumulata viene dispersa mediante urti con particelle che si trovano nelle vicinanze degli elettroni eccitati). Ogni atomo disponendo di un proprio numero di elettroni situati su determinati orbitali, è caratterizzato da specifici spettri di assorbimento atomico, costituiti da una serie di righe di risonanza, la cui intensità è proporzionale alla probabilità della transizione elettronica corrispondente. Ogni transizione, infatti, corrisponde all’assorbimento di una precisa energia e produce una riga spettrale con una caratteristica lunghezza d’onda.")
85
Una linea spettrale chiara o scura in uno spettro altrimenti uniforme e continuo, è la conseguenza di un assorbimento o emissione di fotoni in una stretta gamma di frequenza. Quando un fotone ha l'energia corretta per permettere un cambio nello stato energetico del sistema (nel caso di un atomo è solitamente un salto orbitale di elettrone), il fotone è assorbito. Poi sarà riemesso spontaneamente. La direzione dei nuovi fotoni non sarà in relazione alla direzione del fotone originale. Si osserva, infatti, un calo nell'intensità di luce nella frequenza del fotone incidente, poiché i fotoni riemessi saranno in direzioni diverse dell'originale. Questa è una linea di assorbimento. Le linee d'assorbimento sono estremamente atomo-specifiche, e possono essere usate per identificare facilmente la composizione chimica di tutti i mezzi che la luce può attraversare. Per determinare la quantità di un elemento, si può atomizzare il campione in cui è contenuto, eccitare i suoi atomi con radiazioni di opportuna lunghezza d’onda e misurare l’entità della radiazione assorbita. L’assorbimento, che dipende dal numero di atomi nello stato fondamentale, è direttamente proporzionale alla concentrazione dell’elemento nel campione (se l’atomizzazione viene ottenuto con una fiamma) oppure alla quantità dell’elemento (se l’atomizzazione viene ottenuta con sistemi senza fiamma). In queste condizioni l’assorbimento atomico sia pure in un intervallo di linearità abbastanza ristretto segue una legge analoga alla legge di Beer, descritta per l’assorbimento molecolare. Per un generico elemento eccitato da una radiazione monocromatica i cui atomi siano dispersi in fase gassosa abbiamo che: log Io/I = x b N Dove: x è il coefficiente spettrale di assorbimento atomico b lo spessore dello strato assorbente ( il cammino ottico) N il numero totale di atomi liberi Io = radiazione entrante I = radiazione uscente
, il fotone è assorbito. Poi sarà riemesso spontaneamente. La direzione dei nuovi fotoni non sarà in relazione alla direzione del fotone originale. Si osserva, infatti, un calo nell intensità di luce nella frequenza del fotone incidente, poiché i fotoni riemessi saranno in direzioni diverse dell originale. Questa è una linea di assorbimento. Le linee d assorbimento sono estremamente atomo-specifiche, e possono essere usate per identificare facilmente la composizione chimica di tutti i mezzi che la luce può attraversare. Per determinare la quantità di un elemento, si può atomizzare il campione in cui è contenuto, eccitare i suoi atomi con radiazioni di opportuna lunghezza d’onda e misurare l’entità della radiazione assorbita. L’assorbimento, che dipende dal numero di atomi nello stato fondamentale, è direttamente proporzionale alla concentrazione dell’elemento nel campione (se l’atomizzazione viene ottenuto con una fiamma) oppure alla quantità dell’elemento (se l’atomizzazione viene ottenuta con sistemi senza fiamma). In queste condizioni l’assorbimento atomico sia pure in un intervallo di linearità abbastanza ristretto segue una legge analoga alla legge di Beer, descritta per l’assorbimento molecolare. Per un generico elemento eccitato da una radiazione monocromatica i cui atomi siano dispersi in fase gassosa abbiamo che: log Io/I = x b N. Dove: x è il coefficiente spettrale di assorbimento atomico. b lo spessore dello strato assorbente ( il cammino ottico) N il numero totale di atomi liberi. Io = radiazione entrante. I = radiazione uscente.")
86
Gli spettri a righe vengono oggi impiegati non solo per condurre un’analisi qualitativa, essendo tali spettri caratteristici per ogni atomo (e questo è dovuto alla specifica distribuzione energetica degli orbitali), ma soprattutto per condurre un’analisi quantitativa. ANALISI QUALITATIVA: Spettro caratteristico per ogni atomo, dovuto alla specifica distribuzione energetica degli orbitali ANALISI QUANTITATIVA: Intensità delle righe SPETTROMETRO La sorgente (lampada) di radiazioni è un dispositivo in grado di generare radiazione elettromagnetica, generalmente mediante una scarica elettrica nel vuoto o in un plasma a bassa pressione. Esse generano radiazioni a linee discrete in grado di emettere le linee spettrali di uno o più elementi: di questo tipo le più usate sono le lampade a catodo cavo e lampade a scarica senza elettrodi.
 di radiazioni è un dispositivo in grado di generare radiazione elettromagnetica, generalmente mediante una scarica elettrica nel vuoto o in un plasma a bassa pressione. Esse generano radiazioni a linee discrete in grado di emettere le linee spettrali di uno o più elementi: di questo tipo le più usate sono le lampade a catodo cavo e lampade a scarica senza elettrodi.")
87
Lampade a catodo cavo Contengono un catodo cavo composto dallo stesso elemento da analizzare, o da una sua lega. L'anodo è in genere di tungsteno o nichel. Sia il catodo che l’anodo sono contenti in un cilindro di vetro riempito con un gas inerte (neon o argon) a una pressione di 1 kPa. Se si applica agli elettrodi una differenza di potenziale di V si verifica una parziale ionizzazione del gas di riempimento (ad esempio Argon). Gli ioni positivi Ar+, accelerati dal campo elettrico, urtano il catodo e provocano l’espulsione di atomi di metallo (cioè analita) allo stato fondamentale: questo fenomeno porta alla formazione di atomi vaporizzati, i quali collidono successivamente con altri atomi di Ar+, eccitandosi. In seguito all’eccitazione e al conseguente rilassamento questi atomi emettono energia luminosa come banda spettrale a righe discrete.
 a una pressione di 1 kPa. Se si applica agli elettrodi una differenza di potenziale di V si verifica una parziale ionizzazione del gas di riempimento (ad esempio Argon). Gli ioni positivi Ar+, accelerati dal campo elettrico, urtano il catodo e provocano l’espulsione di atomi di metallo (cioè analita) allo stato fondamentale: questo fenomeno porta alla formazione di atomi vaporizzati, i quali collidono successivamente con altri atomi di Ar+, eccitandosi. In seguito all’eccitazione e al conseguente rilassamento questi atomi emettono energia luminosa come banda spettrale a righe discrete.")
88
Atomizzazione in fiamma
Il dispositivo più utilizzato è un bruciatore a premiscelazione e a flusso laminare. Si basa sulla nebulizzazione di una aliquota di campione (sempre in soluzione) in una fiamma, la quale deve avere abbastanza energia per vaporizzarlo e atomizzarlo. La soluzione viene aspirata attraverso un nebulizzatore pneumatico, la cui velocità di aspirazione non deve superare un certo valore perché altrimenti si avrebbe un abbassamento della temperatura della fiamma, riducendo così l’efficienza dell’atomizzazione e quindi, la sensibilità dell’analisi. Dal nebulizzatore la soluzione viene introdotta come spray di aerosol fine nella camera di premiscelazione dove viene mescolato con una miscela combustibile–comburente. Il gas comburente (rappresentato da aria o protossido di azoto) serve per mantenere viva la fiamma, ma funge anche da gas di trasporto per il nebulizzatore; esso ha quindi due entrate, una nella camera di premiscelazione e una nel nebulizzatore: ciò consente di variare il flusso del nebulizzatore senza variare il flusso alla testata. La miscela ottenuta viene quindi trasferita nel bruciatore vero e proprio dove avviene la volatilizzazione e l'atomizzazione. I materiali con cui sono costruiti i dispositivi del bruciatore sono studiati per essere i più inerti possibili, così il nebulizzatore è di acciaio inox mentre la testata è di titanio.
 in una fiamma, la quale deve avere abbastanza energia per vaporizzarlo e atomizzarlo. La soluzione viene aspirata attraverso un nebulizzatore pneumatico, la cui velocità di aspirazione non deve superare un certo valore perché altrimenti si avrebbe un abbassamento della temperatura della fiamma, riducendo così l’efficienza dell’atomizzazione e quindi, la sensibilità dell’analisi. Dal nebulizzatore la soluzione viene introdotta come spray di aerosol fine nella camera di premiscelazione dove viene mescolato con una miscela combustibile–comburente. Il gas comburente (rappresentato da aria o protossido di azoto) serve per mantenere viva la fiamma, ma funge anche da gas di trasporto per il nebulizzatore; esso ha quindi due entrate, una nella camera di premiscelazione e una nel nebulizzatore: ciò consente di variare il flusso del nebulizzatore senza variare il flusso alla testata. La miscela ottenuta viene quindi trasferita nel bruciatore vero e proprio dove avviene la volatilizzazione e l atomizzazione. I materiali con cui sono costruiti i dispositivi del bruciatore sono studiati per essere i più inerti possibili, così il nebulizzatore è di acciaio inox mentre la testata è di titanio.")
89
Fornetto di grafite Si tratta di un sistema interamente automatizzato, che consente di abbassare notevolmente (1000 volte) i limiti di rivelabilità, inoltre consente di lavorare su aliquote molto piccole di campione. Un piccolo volume di campione (20 – 100 μl) viene introdotto nel tubo di grafite, posto sul cammino ottico della radiazione emessa dalla sorgente. Nel tubo fluisce un gas inerte, che espelle l’aria rendendo l’atmosfera non ossidante e quindi adatta a far rimanere gli atomi del campione allo stato fondamentale. Il tubo (essendo di grafite, cioè un materiale inerte ad alta conducibilità) viene riscaldato elettricamente secondo un programma a tre stadi, condotti a temperature crescenti, che portano in successione a: evaporazione del solvente, incenerimento, atomizzazione La misura di assorbimento viene fatta sui vapori atomici che si liberano rapidamente nello stadio finale del riscaldamento. Il segnale che si ottiene è un picco la cui area (altezza) è direttamente proporzionale alla concentrazione dell’analita presente allo stato atomico nel tubo di grafite.
 i limiti di rivelabilità, inoltre consente di lavorare su aliquote molto piccole di campione. Un piccolo volume di campione (20 – 100 μl) viene introdotto nel tubo di grafite, posto sul cammino ottico della radiazione emessa dalla sorgente. Nel tubo fluisce un gas inerte, che espelle l’aria rendendo l’atmosfera non ossidante e quindi adatta a far rimanere gli atomi del campione allo stato fondamentale. Il tubo (essendo di grafite, cioè un materiale inerte ad alta conducibilità) viene riscaldato elettricamente secondo un programma a tre stadi, condotti a temperature crescenti, che portano in successione a: evaporazione del solvente, incenerimento, atomizzazione. La misura di assorbimento viene fatta sui vapori atomici che si liberano rapidamente nello stadio finale del riscaldamento. Il segnale che si ottiene è un picco la cui area (altezza) è direttamente proporzionale alla concentrazione dell’analita presente allo stato atomico nel tubo di grafite.")
90
Evaporazione del solvente Vaporizzazione dei soluti
Soluzione Fiamma o fornetto Evaporazione del solvente Vaporizzazione dei soluti Dissociazione dei composti Atomi Io I Radiazione monocromatica Radiazione ottenuta Affinché si realizzi il processo di assorbimento atomico è necessario che la specie chimica interessata venga trasformata in atomi individuali. Il campione nebulizzato viene aspirato nel bruciatore dove si miscela con il combustibile e il comburente. Sulla fiamma il solvente evapora ed il sale disciolto fonde, evaporando poi a sua volta. Fino a questo punto l’elemento in esame si trova legato ad un anione, risultando una molecola che non presenta il fenomeno dell’assorbimento atomico. L’energia della fiamma (o del fornetto) permette però la dissociazione in atomi, e la successiva eccitazione. L’energia termica fornita acquista perciò un’importanza fondamentale. La fiamma aria – acetilene è in genere sufficiente per atomizzare la maggior parte degli elementi determinabili via assorbimento atomico. Fiamme meno calde aumentano le interferenze perché l’energia che forniscono è insufficiente all’atomizzazione completa. Il numero di atomi che si formano nella fiamma determina la quantità di luce assorbita. Se un componente qualsiasi presente nella soluzione modifica il comportamento (e il numero) degli atomi che arrivano alla fiamma, le letture di assorbanza possono risultare anche molto diverse.
 permette però la dissociazione in atomi, e la successiva eccitazione. L’energia termica fornita acquista perciò un’importanza fondamentale. La fiamma aria – acetilene è in genere sufficiente per atomizzare la maggior parte degli elementi determinabili via assorbimento atomico. Fiamme meno calde aumentano le interferenze perché l’energia che forniscono è insufficiente all’atomizzazione completa. Il numero di atomi che si formano nella fiamma determina la quantità di luce assorbita. Se un componente qualsiasi presente nella soluzione modifica il comportamento (e il numero) degli atomi che arrivano alla fiamma, le letture di assorbanza possono risultare anche molto diverse.")
91
PIOMBO – Pb Il piombo (Pb) è un metallo di colore grigio chiaro ed opaco. Fonde a temperatura relativamente bassa ( °C), è poco solubile in acqua. Il piombo può essere introdotto nell’organismo anche per via inalatoria sotto forma di polveri e fumi; esso finisce nell’organismo attraverso le vie respiratorie. Alti livelli di esposizione possono provocare effetti biochimici tossici negli esseri umani che comprendono problemi nella sintesi di emoglobina, problemi sui reni, sul tratto gastrointestinale, sui giunti e sul sistema riproduttivo e danneggiamento acuto o cronico del sistema nervoso. L’introduzione delle benzine a bassissimo contenuto di piombo, ha ridotto considerevolmente il contributo delle emissioni da trasporto all’inquinamento da piombo. Dal 1° gennaio 2005 il metodo di riferimento per il campionamento del piombo è quello utilizzato per il PM10. Metodo di analisi: ISO 9855:1993 – Determinazione del contenuto di piombo negli areosols raccolti su filtro – spettroscopia ad assorbimento atomico Il metodo analitico prevede per l’estrazione del contenuto di piombo la mineralizzazione del campione con acido nitrico e la determinazione finale con spettrometria ad assorbimento atomico.
, è poco solubile in acqua. Il piombo può essere introdotto nell’organismo anche per via inalatoria. sotto forma di polveri e fumi; esso finisce nell’organismo attraverso le vie respiratorie. Alti livelli di esposizione possono provocare effetti biochimici tossici negli esseri umani che comprendono problemi nella sintesi di emoglobina, problemi sui reni, sul tratto gastrointestinale, sui giunti e sul sistema riproduttivo e danneggiamento acuto o cronico del sistema nervoso. L’introduzione delle benzine a bassissimo contenuto di piombo, ha ridotto considerevolmente il contributo delle emissioni da trasporto all’inquinamento da piombo. Dal 1° gennaio 2005 il metodo di riferimento per il campionamento del piombo è quello utilizzato per il PM10. Metodo di analisi: ISO 9855:1993 – Determinazione del contenuto di piombo negli areosols raccolti su filtro – spettroscopia ad assorbimento atomico. Il metodo analitico prevede per l’estrazione del contenuto di piombo la mineralizzazione del campione con acido nitrico e la determinazione finale con spettrometria ad assorbimento atomico.")
92
Mineralizzazione del filtro
Il filtro va collocato in un recipiente di vetro a fondo piano nel quale si aggiungono 3 mL di acido nitrico concentrato (HNO3 = 1.40 g/mL) con concentrazione di piombo uguale o inferiore a 70 mg/l. Si scalda sotto cappa aspirante su piastra riscaldante mantenuta a temperatura non superiore a 200 °C, portando quasi a secco. Si completa la mineralizzazione ripetendo il trattamento per altre due volte con 2 mL di acido nitrico per volta. Il residuo ottenuto è trattato con 1 mL di acido nitrico e diluito conn 3 mL di acqua distillata. Si procede alla determinazione del piombo sia nella soluzione del campione, che nella prova in bianco, usando lo spettrofotometro di assorbimento atomico. Si legge l'assorbanza a nm.
 con concentrazione di piombo uguale o inferiore a 70 mg/l. Si scalda sotto cappa aspirante su piastra riscaldante mantenuta a temperatura non superiore a 200 °C, portando quasi a secco. Si completa la mineralizzazione ripetendo il trattamento per altre due volte con 2 mL di acido nitrico per volta. Il residuo ottenuto è trattato con 1 mL di acido nitrico e diluito conn 3 mL di acqua distillata. Si procede alla determinazione del piombo sia nella soluzione del campione, che nella prova in bianco, usando lo spettrofotometro di assorbimento atomico. Si legge l assorbanza a nm.")
Presentazioni simili

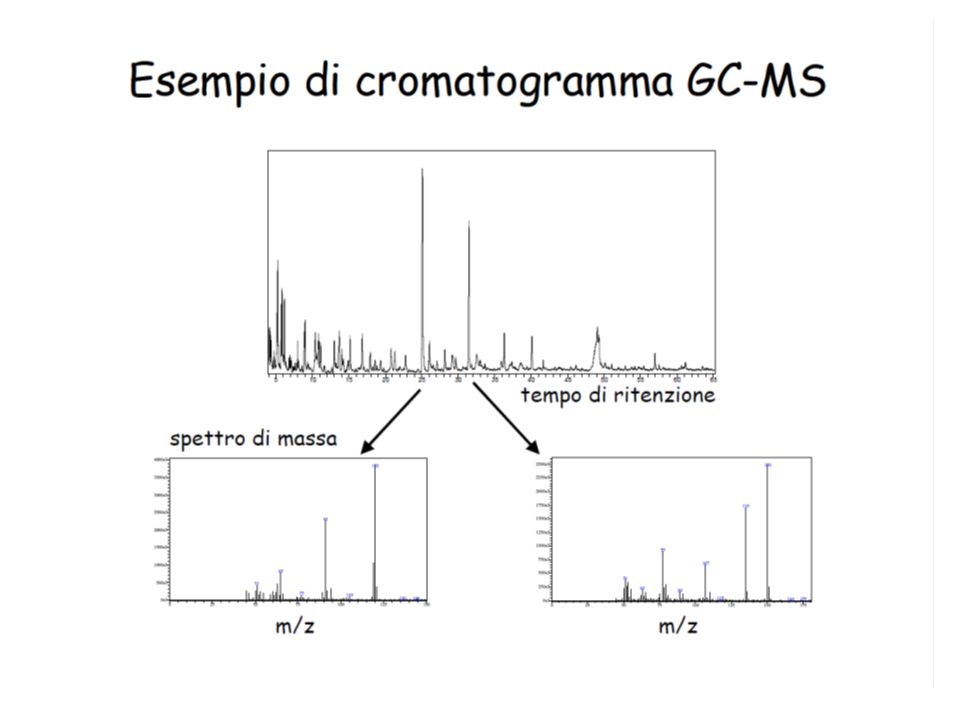
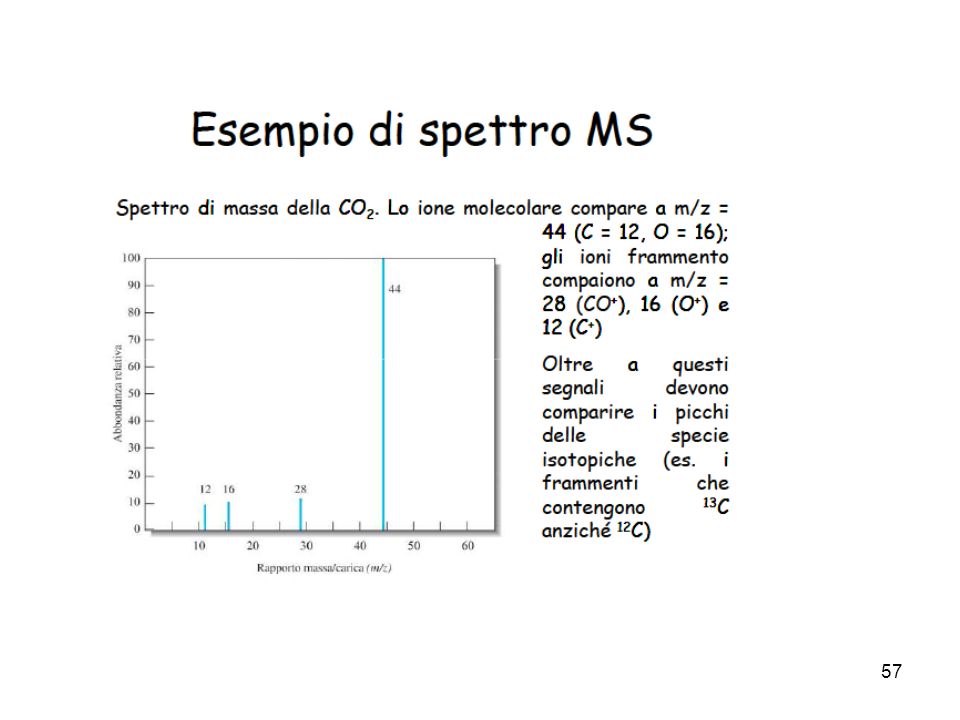








>")

 tra la radiazione.>")








