Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Stabilizzatori dell’umore

2
Journal of Americal Medical Association (1910)
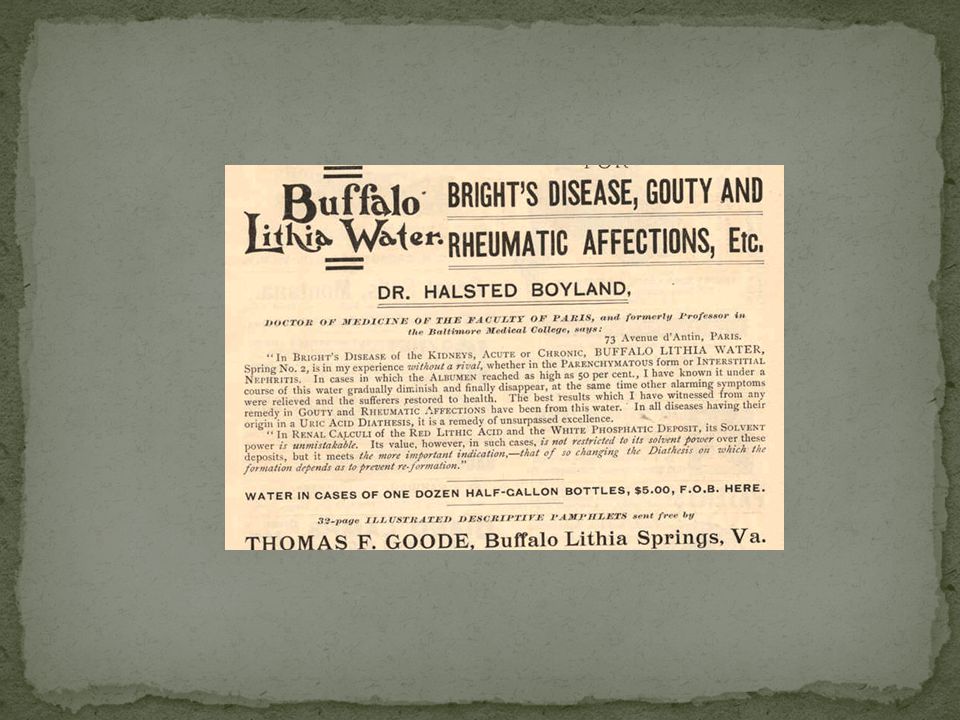
“gotta, reumatismo, diatesi all’iperuricemia, stipsi acuta e cronica, torpore epatico, obesità, malattia di Bright, albuminuria della gravidanza, asma, incontinenza urinaria, calcolosi, cistite, disturbi urogenitali, nevralgia e lombalgia…e anche un buon antimalarico”. Charles Mitchell

6
Farmaci stabilizzatori dell’umore
1949 efficacia dei sali di Litio nel trattamento dell’eccitamento psicotico (Cade, Medical Journal of Australia) 1954 primo studio controllato sull’efficacia antimaniacale (Schou) Negli anni 60 Shepherd e Lewis definiscono il litio un “pericoloso nonsense”e, in seguito, un “mito terapeutico” basato su “seri difetti metodologici” e “affermazioni false” Nel 1970 Lancet pubblicò uno studio in doppio cieco sulla efficacia del Litio nella profilassi della psicosi maniaco- depressiva (Schou et al) e divenne lo stabilizzante dell’umore di prima scelta La scoperta dell’attività antimaniacale del Litio fu casuale, in quanto il suo effetto sedativo sull’animale (porcellini d’india) fu evidenziato cercando di stabilizzare l’urea come urato di Litio per dimostrare gli effetti tossici di quest’ultima nella mania Fu approvato negli USA solo 1970; sostanzialmente due fattori ne ritardarono l’impiego: la cardiotossicità a dosaggi elevati (fino a 4/6 grammi die) e lo scarso interesse dell’industria farmaceutica per un composto dal costo irrisorio, senza alcun brevetto e di facile preparazione. In Italia iniziò ad essere introdotto solo agli inizi degli anni 70. Storia del litio e del suo impiego in medicina ed in psichiatria (prima parte) A cura di Giuseppe Ruffolo La storia ha inizio nel 1817 quando Johan Arfwedson scoprì un nuovo metallo alcalino che Jons Jacob Berzelius denominò “lithion”. L’interesse per l’impiego del litio in campo medico risale alla metà del 1800 quando A. Lipowitz ed A. Ure descrissero la sua proprietà di dissolvere “in vitro” i cristalli di acido urico; sulla base di questa osservazione si pensò che esso potesse essere utile nel trattamento della gotta. Il medico inglese Sir A.B. Garrod, convinto sostenitore dell’impiego del litio nel trattamento di tale patologia, si adoperò per stabilirne i dosaggi terapeutici e, per primo, ne descrisse i principali effetti collaterali. Furono tuttavia gli scritti di Alexander Haig a rendere il litio popolare; Haig sosteneva che diverse patologie quali angina pectoris, asma, artrite, depressione, mal di testa, ipertensione ed epilessia potessero essere causate da una disfunzione metabolica a carico dell’acido urico: il litio, alla luce delle sue proprietà favorenti la dissoluzione dei cristalli di urato, avrebbe dunque potuto svolgere un ruolo importante nel trattamento di queste patologie. Negli anni successivi, le affermazioni relative alle potenzialità terapeutiche del litio si moltiplicarono. Nel Garrod formulò l’ipotesi secondo la quale i disturbi dell’umore potevano essere la risultante dell’accumulo di urati a livello cerebrale: il litio dunque, alla luce delle sue proprietà favorenti la dissoluzione di tali cristalli “in vitro”, sarebbe potuto essere un utile strumento terapeutico. Nel 1910, in un numero del Journal of Americal Medical Association, Charles Mitchell, un medico che si occupava della produzione di medicinali, affermò che il suo “lassativo alcalino a base di sali di litio” era indicato per il trattamento di “gotta, reumatismo, diatesi all’iperuricemia, stipsi acuta e cronica, torpore epatico, obesità, malattia di Bright, albuminuria della gravidanza, asma, incontinenza urinaria, calcolosi, cistite, disturbi urogenitali, nevralgia e lombalgia”; inoltre, secondo Mitchell, “il litio era anche un buon antimalarico”. Questo crescente interesse per il litio stimolò fortemente la diffusione e la commercializzazione di prodotti che lo contenessero, in particolare acque minerali (Lithia Water) e birra (Lithia Beer). La “moda” delle bevande a base di litio, dopo un iniziale successo, andò tuttavia spegnendosi progressivamente; a tale declino contribuì in maniera significativa una ricerca effettuata negli Stati Uniti i cui risultati misero in evidenza che sarebbe stato necessario bere diverse migliaia di litri al giorno di Buffalo Lithia Water (un’acqua minerale commercializzata a quell’epoca) per ottenere una singola dose giornaliera terapeutica di litio. A partire dalla fine del 1940 l’interesse per il litio s’incentrò particolarmente sul suo impiego come sostituto del sale da cucina (sotto forma di cloridrato) in pazienti con problemi cardiovascolari tuttavia, l’ingestione di significative quantità di sali di litio in soggetti con problemi medici di tal genere, peraltro con funzionalità renale spesso compromessa e in terapia con farmaci diuretici, finì col provocare alcuni fenomeni d’intossicazione. Nel corso del 1949 furono infatti pubblicati diversi “reports” che descrivevano casi di avvelenamento dovuti ad intossicazione da litio cloridrato. Il verificarsi di questi eventi condizionò significativamente la diffusione del litio, sia in campo medico che psichiatrico. La brillante intuizione di John Cade, che ha aperto la strada alla sperimentazione del litio nel campo delle malattie mentali, risale al 1949, lo stesso anno in cui si resero evidenti i risultati della sua tossicità; essa precede quindi di ben vent’anni l’approvazione del litio nel trattamento delle fasi di eccitazione maniacale da parte della Food and Drug Administration americana (1970). Nella seconda parte di questa rassegna percorreremo le principali tappe attraverso le quali il litio si è affermato come strumento “epocale” per il trattamento del Disturbo Bipolare nelle sue diverse fasi [depressione, (ipo)mania, profilassi delle ricadute]. Storia del litio e del suo impiego in medicina ed in psichiatria (seconda ed ultima parte) John Cade (allora sconosciuto psichiatra australiano) lavorava, con i pochissimi mezzi che gli erano messi a disposizione, in un piccolo ospedale per pazienti psichiatrici cronici; la sua storia dimostra come le scoperte più straordinarie possano talora scaturire da ipotesi iniziali non del tutto corrette e da brillanti intuizioni successive. Era il 1949 quando Cade, presupponendo che la mania fosse causata dall’eccesso di una qualche sostanza fisiologicamente presente nell’organismo, osservò che l’iniezione intra-peritoneale di urina di pazienti maniaci in porcellini d’india ne provocava la morte molto più rapidamente di quanto non facesse quella di soggetti normali, schizofrenici oppure melanconici. Cade identificò nell’urea la sostanza tossica responsabile di tale effetto e, nel tentativo di stabilire quanto l’acido urico influenzasse la tossicità dell’urea, somministrò ai porcellini d’india litio urato (il più solubile fra gli urati) unitamente ad una soluzione contenente urea. Successivamente, alfine di determinare se i sali di litio avessero di per se stessi un qualche effetto percettibile, iniettò carbonato di litio negli animali da esperimento osservando che essi, dopo circa due ore, seppur lucidi, apparivano fiacchi e poco responsivi agli stimoli, per poi riprendere la loro normale attività. Può essere ipotizzato che la “procurata” docilità dei porcellini d’india notata da Cade fosse stata dovuta all’effetto tossico dell’iniezione di litio carbonato tuttavia egli, sulla base del presunto effetto sedativo osservato, decise di intraprendere uno studio naturalistico con lo scopo di valutare quali fossero gli effetti del litio nei soggetti in fase maniacale. Il primo dei dieci pazienti maniaci selezionati da Cade era un uomo di 51 anni in uno stato d’eccitamento maniacale oramai cronico, refrattario alle terapie allora disponibili e destinato a rimanere nel reparto per cronici dove Cade stesso lavorava per tutto il resto della sua vita. Il trattamento con sali di litio venne iniziato il 29 Marzo del 1948; già dopo pochi giorni i risultati apparvero assolutamente sorprendenti oltre che inaspettati: il paziente era più stabile, meno disinibito, meno distraibile e meno aggressivo. I miglioramenti continuarono nelle settimane successive e furono tali da consentirgli in seguito di riprendere l’attività lavorativa che da anni aveva dovuto abbandonare. Cade rilevò risultati ugualmente favorevoli negli altri nove pazienti descritti nel suo, ormai storico, studio osservazionale. Il nostro racconto sul progressivo affermarsi del litio come terapia d’elezione della malattia maniaco-depressiva ci porta adesso a diverse migliaia di chilometri di distanza, dall’Australia, patria di Cade, all’Europa, in Danimarca. Era il 1951 quando un giovane e brillante ricercatore danese, Mogens Schou, rimasto colpito dalla pubblicazione di Cade sull’effetto antimaniacale del litio, decise di approfondire tale argomento ricorrendo a procedure improntate ad un maggior rigore scientifico. A tale scopo Schou concepì, in collaborazione con i suoi colleghi Strömgren, Juel-Nielsen e Voldby, una sperimentazione, parte in aperto e parte randomizzata e controllata verso placebo, sull’impiego del litio in pazienti maniacali; i risultati della ricerca confermarono pienamente e con solide basi scientifiche la correttezza di quanto Cade aveva osservato due anni prima. Tuttavia, come spesso accade, tutto ciò che è “novità”, discostandosi da quello che è il “sentire” prevalente, può faticare ad affermarsi. Mogens Schou inviò il suo manoscritto ad Eliot Slater, allora “assistant editor” del Journal of Mental Science (precursore del attuale Britisch Journal of Psychiatry) che non lo ritenne interessante per la pubblicazione in quanto l’impiego del litio in psichiatria era allora praticamente sconosciuto tuttavia, lo stesso Slater, gli suggerì di sottoporlo all’attenzione di una rivista disposta a pubblicare contributi meno “usuali”; l’articolo fu così inviato al Journal of Neurology, Neurosurgery and Psychiatry che lo pubblicò nel 1954. Nei dieci anni successivi Mogens Schou ed i suoi colleghi Alec Coppen, Nathan Kline e Sam Gershon impegnarono la maggior parte delle loro energie nella difficile battaglia volta a far “conoscere” e “riconoscere” il ruolo primario del litio nel trattamento degli episodi maniacali tuttavia, la Food and Drug Administration americana, lo ha approvato ufficialmente come terapia elettiva degli stati d’eccitamento maniacale diversi anni più tardi, nel 1970. Nel corso degli anni ’60 del secolo scorso, indipendentemente l’uno dall’altro, Schou ed altri due psichiatri, Hartigan e Baastrup, notarono che il litio appariva dotato anche di un effetto profilattico sulle ricorrenze della malattia maniaco-depressiva. Baastrup e Schou decisero di riunire le loro forze e condussero il primo di una serie di studi che confermarono pienamente quelle iniziali, sporadiche, osservazioni. Dunque, il litio era sì efficace sul controllo degli episodi maniacali, ma era dotato anche di un effetto preventivo sulle ricadute, depressive, maniacali, ipomaniacali. Inutile sottolineare che queste osservazioni avevano il sapore di una vera e propria svolta nella cura della malattia maniaco-depressiva. L’ipotesi di Schou e Baastrup sull’efficacia profilattica del litio trovò tuttavia notevole resistenza alla diffusione fra gli psichiatri britannici; fra i più critici Michael Schepard secondo il quale il litio era “un pericoloso non senso”, un “mito terapeutico” basato su “affermazioni spurie” e su “seri difetti metodologici”. Le motivazioni di tali violenti attacchi furono forse dovute al fatto che la psichiatria britannica di quegli anni era dominata dalle teorie di Aubrey Lewis il quale non operava distinzione alcuna fra depressione psicogena ed endogena per cui è ipotizzabile che fra gli psichiatri britannici si fosse diffuso il timore che, se il litio fosse stato “raccomandato” nella terapia della depressione, il risultato sarebbe potuto essere una prescrizione troppo generalizzata che avrebbe potuto aumentare pericolosamente i rischi connessi alla sua potenziale tossicità (uno stigma, quest’ultimo che per la verità anche tuttora, seppur in parte, persiste). Dunque, anche questa volta, il percorso è stato lungo e difficile tuttavia, dopo molti anni, riconosciuti esperti nel campo dello studio della malattia maniaco-depressiva come Fred Goodwin e Kay Jamison (1990) hanno affermato che la scoperta della terapia profilattica con sali di litio ha rappresentato “uno dei più importanti progressi della psichiatria moderna”. 6
 1954 primo studio controllato sull’efficacia antimaniacale (Schou) Negli anni 60 Shepherd e Lewis definiscono il litio un pericoloso nonsense e, in seguito, un mito terapeutico basato su seri difetti metodologici e affermazioni false Nel 1970 Lancet pubblicò uno studio in doppio cieco sulla efficacia del Litio nella profilassi della psicosi maniaco- depressiva (Schou et al) e divenne lo stabilizzante dell’umore di prima scelta. La scoperta dell’attività antimaniacale del Litio fu casuale, in quanto il suo effetto sedativo sull’animale (porcellini d’india) fu evidenziato cercando di stabilizzare l’urea come urato di Litio per dimostrare gli effetti tossici di quest’ultima nella mania. Fu approvato negli USA solo 1970; sostanzialmente due fattori ne ritardarono l’impiego: la cardiotossicità a dosaggi elevati (fino a 4/6 grammi die) e lo scarso interesse dell’industria farmaceutica per un composto dal costo irrisorio, senza alcun brevetto e di facile preparazione. In Italia iniziò ad essere introdotto solo agli inizi degli anni 70. Storia del litio e del suo impiego in medicina ed in psichiatria (prima parte) A cura di Giuseppe Ruffolo. La storia ha inizio nel 1817 quando Johan Arfwedson scoprì un nuovo metallo alcalino che Jons Jacob Berzelius denominò lithion . L’interesse per l’impiego del litio in campo medico risale alla metà del 1800 quando A. Lipowitz ed A. Ure descrissero la sua proprietà di dissolvere in vitro i cristalli di acido urico; sulla base di questa osservazione si pensò che esso potesse essere utile nel trattamento della gotta. Il medico inglese Sir A.B. Garrod, convinto sostenitore dell’impiego del litio nel trattamento di tale patologia, si adoperò per stabilirne i dosaggi terapeutici e, per primo, ne descrisse i principali effetti collaterali. Furono tuttavia gli scritti di Alexander Haig a rendere il litio popolare; Haig sosteneva che diverse patologie quali angina pectoris, asma, artrite, depressione, mal di testa, ipertensione ed epilessia potessero essere causate da una disfunzione metabolica a carico dell’acido urico: il litio, alla luce delle sue proprietà favorenti la dissoluzione dei cristalli di urato, avrebbe dunque potuto svolgere un ruolo importante nel trattamento di queste patologie. Negli anni successivi, le affermazioni relative alle potenzialità terapeutiche del litio si moltiplicarono. Nel 1876 Garrod formulò l’ipotesi secondo la quale i disturbi dell’umore potevano essere la risultante dell’accumulo di urati a livello cerebrale: il litio dunque, alla luce delle sue proprietà favorenti la dissoluzione di tali cristalli in vitro , sarebbe potuto essere un utile strumento terapeutico. Nel 1910, in un numero del Journal of Americal Medical Association, Charles Mitchell, un medico che si occupava della produzione di medicinali, affermò che il suo lassativo alcalino a base di sali di litio era indicato per il trattamento di gotta, reumatismo, diatesi all’iperuricemia, stipsi acuta e cronica, torpore epatico, obesità, malattia di Bright, albuminuria della gravidanza, asma, incontinenza urinaria, calcolosi, cistite, disturbi urogenitali, nevralgia e lombalgia ; inoltre, secondo Mitchell, il litio era anche un buon antimalarico . Questo crescente interesse per il litio stimolò fortemente la diffusione e la commercializzazione di prodotti che lo contenessero, in particolare acque minerali (Lithia Water) e birra (Lithia Beer). La moda delle bevande a base di litio, dopo un iniziale successo, andò tuttavia spegnendosi progressivamente; a tale declino contribuì in maniera significativa una ricerca effettuata negli Stati Uniti i cui risultati misero in evidenza che sarebbe stato necessario bere diverse migliaia di litri al giorno di Buffalo Lithia Water (un’acqua minerale commercializzata a quell’epoca) per ottenere una singola dose giornaliera terapeutica di litio. A partire dalla fine del 1940 l’interesse per il litio s’incentrò particolarmente sul suo impiego come sostituto del sale da cucina (sotto forma di cloridrato) in pazienti con problemi cardiovascolari tuttavia, l’ingestione di significative quantità di sali di litio in soggetti con problemi medici di tal genere, peraltro con funzionalità renale spesso compromessa e in terapia con farmaci diuretici, finì col provocare alcuni fenomeni d’intossicazione. Nel corso del 1949 furono infatti pubblicati diversi reports che descrivevano casi di avvelenamento dovuti ad intossicazione da litio cloridrato. Il verificarsi di questi eventi condizionò significativamente la diffusione del litio, sia in campo medico che psichiatrico. La brillante intuizione di John Cade, che ha aperto la strada alla sperimentazione del litio nel campo delle malattie mentali, risale al 1949, lo stesso anno in cui si resero evidenti i risultati della sua tossicità; essa precede quindi di ben vent’anni l’approvazione del litio nel trattamento delle fasi di eccitazione maniacale da parte della Food and Drug Administration americana (1970). Nella seconda parte di questa rassegna percorreremo le principali tappe attraverso le quali il litio si è affermato come strumento epocale per il trattamento del Disturbo Bipolare nelle sue diverse fasi [depressione, (ipo)mania, profilassi delle ricadute]. Storia del litio e del suo impiego in medicina ed in psichiatria (seconda ed ultima parte) John Cade (allora sconosciuto psichiatra australiano) lavorava, con i pochissimi mezzi che gli erano messi a disposizione, in un piccolo ospedale per pazienti psichiatrici cronici; la sua storia dimostra come le scoperte più straordinarie possano talora scaturire da ipotesi iniziali non del tutto corrette e da brillanti intuizioni successive. Era il 1949 quando Cade, presupponendo che la mania fosse causata dall’eccesso di una qualche sostanza fisiologicamente presente nell’organismo, osservò che l’iniezione intra-peritoneale di urina di pazienti maniaci in porcellini d’india ne provocava la morte molto più rapidamente di quanto non facesse quella di soggetti normali, schizofrenici oppure melanconici. Cade identificò nell’urea la sostanza tossica responsabile di tale effetto e, nel tentativo di stabilire quanto l’acido urico influenzasse la tossicità dell’urea, somministrò ai porcellini d’india litio urato (il più solubile fra gli urati) unitamente ad una soluzione contenente urea. Successivamente, alfine di determinare se i sali di litio avessero di per se stessi un qualche effetto percettibile, iniettò carbonato di litio negli animali da esperimento osservando che essi, dopo circa due ore, seppur lucidi, apparivano fiacchi e poco responsivi agli stimoli, per poi riprendere la loro normale attività. Può essere ipotizzato che la procurata docilità dei porcellini d’india notata da Cade fosse stata dovuta all’effetto tossico dell’iniezione di litio carbonato tuttavia egli, sulla base del presunto effetto sedativo osservato, decise di intraprendere uno studio naturalistico con lo scopo di valutare quali fossero gli effetti del litio nei soggetti in fase maniacale. Il primo dei dieci pazienti maniaci selezionati da Cade era un uomo di 51 anni in uno stato d’eccitamento maniacale oramai cronico, refrattario alle terapie allora disponibili e destinato a rimanere nel reparto per cronici dove Cade stesso lavorava per tutto il resto della sua vita. Il trattamento con sali di litio venne iniziato il 29 Marzo del 1948; già dopo pochi giorni i risultati apparvero assolutamente sorprendenti oltre che inaspettati: il paziente era più stabile, meno disinibito, meno distraibile e meno aggressivo. I miglioramenti continuarono nelle settimane successive e furono tali da consentirgli in seguito di riprendere l’attività lavorativa che da anni aveva dovuto abbandonare. Cade rilevò risultati ugualmente favorevoli negli altri nove pazienti descritti nel suo, ormai storico, studio osservazionale. Il nostro racconto sul progressivo affermarsi del litio come terapia d’elezione della malattia maniaco-depressiva ci porta adesso a diverse migliaia di chilometri di distanza, dall’Australia, patria di Cade, all’Europa, in Danimarca. Era il 1951 quando un giovane e brillante ricercatore danese, Mogens Schou, rimasto colpito dalla pubblicazione di Cade sull’effetto antimaniacale del litio, decise di approfondire tale argomento ricorrendo a procedure improntate ad un maggior rigore scientifico. A tale scopo Schou concepì, in collaborazione con i suoi colleghi Strömgren, Juel-Nielsen e Voldby, una sperimentazione, parte in aperto e parte randomizzata e controllata verso placebo, sull’impiego del litio in pazienti maniacali; i risultati della ricerca confermarono pienamente e con solide basi scientifiche la correttezza di quanto Cade aveva osservato due anni prima. Tuttavia, come spesso accade, tutto ciò che è novità , discostandosi da quello che è il sentire prevalente, può faticare ad affermarsi. Mogens Schou inviò il suo manoscritto ad Eliot Slater, allora assistant editor del Journal of Mental Science (precursore del attuale Britisch Journal of Psychiatry) che non lo ritenne interessante per la pubblicazione in quanto l’impiego del litio in psichiatria era allora praticamente sconosciuto tuttavia, lo stesso Slater, gli suggerì di sottoporlo all’attenzione di una rivista disposta a pubblicare contributi meno usuali ; l’articolo fu così inviato al Journal of Neurology, Neurosurgery and Psychiatry che lo pubblicò nel Nei dieci anni successivi Mogens Schou ed i suoi colleghi Alec Coppen, Nathan Kline e Sam Gershon impegnarono la maggior parte delle loro energie nella difficile battaglia volta a far conoscere e riconoscere il ruolo primario del litio nel trattamento degli episodi maniacali tuttavia, la Food and Drug Administration americana, lo ha approvato ufficialmente come terapia elettiva degli stati d’eccitamento maniacale diversi anni più tardi, nel Nel corso degli anni ’60 del secolo scorso, indipendentemente l’uno dall’altro, Schou ed altri due psichiatri, Hartigan e Baastrup, notarono che il litio appariva dotato anche di un effetto profilattico sulle ricorrenze della malattia maniaco-depressiva. Baastrup e Schou decisero di riunire le loro forze e condussero il primo di una serie di studi che confermarono pienamente quelle iniziali, sporadiche, osservazioni. Dunque, il litio era sì efficace sul controllo degli episodi maniacali, ma era dotato anche di un effetto preventivo sulle ricadute, depressive, maniacali, ipomaniacali. Inutile sottolineare che queste osservazioni avevano il sapore di una vera e propria svolta nella cura della malattia maniaco-depressiva. L’ipotesi di Schou e Baastrup sull’efficacia profilattica del litio trovò tuttavia notevole resistenza alla diffusione fra gli psichiatri britannici; fra i più critici Michael Schepard secondo il quale il litio era un pericoloso non senso , un mito terapeutico basato su affermazioni spurie e su seri difetti metodologici . Le motivazioni di tali violenti attacchi furono forse dovute al fatto che la psichiatria britannica di quegli anni era dominata dalle teorie di Aubrey Lewis il quale non operava distinzione alcuna fra depressione psicogena ed endogena per cui è ipotizzabile che fra gli psichiatri britannici si fosse diffuso il timore che, se il litio fosse stato raccomandato nella terapia della depressione, il risultato sarebbe potuto essere una prescrizione troppo generalizzata che avrebbe potuto aumentare pericolosamente i rischi connessi alla sua potenziale tossicità (uno stigma, quest’ultimo che per la verità anche tuttora, seppur in parte, persiste). Dunque, anche questa volta, il percorso è stato lungo e difficile tuttavia, dopo molti anni, riconosciuti esperti nel campo dello studio della malattia maniaco-depressiva come Fred Goodwin e Kay Jamison (1990) hanno affermato che la scoperta della terapia profilattica con sali di litio ha rappresentato uno dei più importanti progressi della psichiatria moderna . 6.")
7
Litio interferisce con il sistema del fosfatidilinositolo (PIP2), attraverso l’inibizione dell’enzima inositolo monofosfatasi, riducendo la disponibilità di inositolo libero. Questo ha un effetto sull’azione di molti neurotrasmettitori, la cui trasduzione del segnale è collegata al ciclo del PIP2 interferisce inoltre con la protein-chinasi C, secondo messaggero importante nella trasduzione del segnale Inibisce il GSK-3 (glycogen synthase kinase) che è un importante regolatore della trasduzione del segnale E’ stato visto che l’inositolo esogeno può alleviare la depressione ed è stata proposta una complessa relazione “a pendolo” tra Litio e inositolo Lithium is widely used to treat bipolar disorder, but its mechanism of action in this disorder is unknown. Lithium directly inhibits glycogen synthase kinase-3 (GSK3), a critical regulator of multiple signal transduction pathways. Inhibition of GSK3 provides a compelling explanation for many of the known effects of lithium, including effects on early development and insulin signaling/ glycogen synthesis
, attraverso l’inibizione dell’enzima inositolo monofosfatasi, riducendo la disponibilità di inositolo libero. Questo ha un effetto sull’azione di molti neurotrasmettitori, la cui trasduzione del segnale è collegata al ciclo del PIP2. interferisce inoltre con la protein-chinasi C, secondo messaggero importante nella trasduzione del segnale. Inibisce il GSK-3 (glycogen synthase kinase) che è un importante regolatore della trasduzione del segnale. E’ stato visto che l’inositolo esogeno può alleviare la depressione ed è stata proposta una complessa relazione a pendolo tra Litio e inositolo. Lithium is widely used to treat bipolar disorder, but its mechanism of action in this disorder is. unknown. Lithium directly inhibits glycogen synthase kinase-3 (GSK3), a critical regulator of. multiple signal transduction pathways. Inhibition of GSK3 provides a compelling explanation for. many of the known effects of lithium, including effects on early development and insulin signaling/ glycogen synthesis.")
8
Ciclo dell’inositolo e ruolo del litio
Fluido extracellulare A Membrana cellulare PI PIP + PIP 2 PKC R PLC G CMP-PA + DAG Abbreviazioni: A=agonista del recettore; Ca2+=ioni calcio; CMP=Citidina monofosfato; DAG=diacilglicerolo; G=proteina G; I=inositolo; I-1-P =mioinositolo 1-fosfato; I-3-P =mioinositolo 3-fosfato; I-4-P =mioinositolo 4-fosfato; IMP=inositolo monofosfatasi; IP3=mioinositolo 1,4,5-trifosfato; IP3R=recettore dell’inositolo trifosfato; Li+=ioni litio; PA=fosfotidato; PI=fosfatidil inositolo; PIP2=fosfatidilinositolo 4,5-difosfato; PKC=proteina chinasi C; PLC=fosfolipasi C; R=recettore; SER=reticolo endoplasmico liscio Lenox RH, Watson DG. Lithium and the brain: a psychopharmacological strategy to a molecular basis for manic depressive illness. Clin Chem 1994;40(2): Department of Psychiatry, University of Vermont College of Medicine, Burlington Lithium, an effective treatment for mania and the prevention of recurrent episodes of both mania and depression in patients with manic depressive illness, exerts multiple biochemical effects. However, any clinically relevant site of action of lithium must occur at therapeutic concentrations attained in the brain of patients and must account for the lag period accompanying onset of action as well as effects persisting beyond discontinuation of treatment. This monovalent cation acts as a potent uncompetitive inhibitor in the receptor-coupled breakdown of inositol phospholipids, resulting in a relative depletion of inositol and an alteration in the generation of diacylglycerol, an endogenous activator of protein kinase C. In our laboratory, we are examining the action of chronically administered lithium on posttranslational modification of specific phosphoproteins involved in regulating signal transduction in the brain. We have found that chronic, but not acute, administration of lithium in rats markedly reduces a major phosphoprotein substrate of protein kinase C in the hippocampus, an effect that persists beyond the cessation of lithium treatment. This protein, myristoylated alanine-rich C kinase substrate ("MARCKS"), is implicated in synaptic neurotransmission, calcium regulation, and cytoskeletal restructuring. These findings have relevance for the long-term action of lithium in stabilizing an underlying dysregulation in the brain and may move us closer to formulating a molecular basis of manic depressive illness. CMP IP3 PA I-4-P IMP I-1-P I I-3-P – Citosol Li + Beaulieau and Caron, 2008
: Department of Psychiatry, University of Vermont College of Medicine, Burlington Lithium, an effective treatment for mania and the prevention of recurrent episodes of both mania and depression in patients with manic depressive illness, exerts multiple biochemical effects. However, any clinically relevant site of action of lithium must occur at therapeutic concentrations attained in the brain of patients and must account for the lag period accompanying onset of action as well as effects persisting beyond discontinuation of treatment. This monovalent cation acts as a potent uncompetitive inhibitor in the receptor-coupled breakdown of inositol phospholipids, resulting in a relative depletion of inositol and an alteration in the generation of diacylglycerol, an endogenous activator of protein kinase C. In our laboratory, we are examining the action of chronically administered lithium on posttranslational modification of specific phosphoproteins involved in regulating signal transduction in the brain. We have found that chronic, but not acute, administration of lithium in rats markedly reduces a major phosphoprotein substrate of protein kinase C in the hippocampus, an effect that persists beyond the cessation of lithium treatment. This protein, myristoylated alanine-rich C kinase substrate ( MARCKS ), is implicated in synaptic neurotransmission, calcium regulation, and cytoskeletal restructuring. These findings have relevance for the long-term action of lithium in stabilizing an underlying dysregulation in the brain and may move us closer to formulating a molecular basis of manic depressive illness. CMP. IP3. PA. I-4-P. IMP. I-1-P. I. I-3-P. – Citosol. Li. + Beaulieau and Caron,")
9
The receptor-2nd messenger-transcription factors cascade
Neurotransmitters First messengers RECEPTORS G PROTEINS Coupling factors Ca2+ DIACYLGLYCEROL IP cAMP cGMP Second messengers PROTEIN KINASES Si ipotizza che alla base della plasticità neurale indotta dai farmaci psicotropi, alla quale viene accreditato il beneficio sintomatologico, si verifichi una modificazione dell’espressione del DNA neuronale in alcune aree cerebrali. Verrebbe quindi stimolata la trascrizione di alcuni geni ed in questo modo si indurrebbero alterazioni funzionali del neurone. I geni immediati precoci (IEG, immediatly early genes) sono rapidamente e transitoriamente attivati a livello del SNC attraverso stimoli diversi, quali i farmaci che inducono la formazione dell’cAMP o dell’inositolotrifosfato o l’incremento del calcio intraneuronale[i]. Tale dato ha fatto ipotizzare che l’espressione degli IEG c-fos e jun possa essere considerata un potenziale marcatore dell’attivazione del neurone[ii]. [i] Morgan JI, Curran T. Stimulus-transcription coupling in neurons: role of cellular immediate-early genes. Trends in Neurosciences, 1989; [ii] Sagar SM, Sharp FR, Curran T. Expression of c-Fos protein in brain: metabolic mapping at the cellular level. Science,1988; 240: CREB-LIKE TRANSCRIPTION FACTORS Third messengers FOS-LIKE TRANSCRIPTION FACTORS OTHER GENES Fourth messengers
 sono rapidamente e transitoriamente attivati a livello del SNC attraverso stimoli diversi, quali i farmaci che inducono la formazione dell’cAMP o dell’inositolotrifosfato o l’incremento del calcio intraneuronale[i]. Tale dato ha fatto ipotizzare che l’espressione degli IEG c-fos e jun possa essere considerata un potenziale marcatore dell’attivazione del neurone[ii]. [i] Morgan JI, Curran T. Stimulus-transcription coupling in neurons: role of cellular immediate-early genes. Trends in Neurosciences, 1989; [ii] Sagar SM, Sharp FR, Curran T. Expression of c-Fos protein in brain: metabolic mapping at the cellular level. Science,1988; 240: CREB-LIKE TRANSCRIPTION FACTORS. Third messengers. FOS-LIKE TRANSCRIPTION FACTORS. OTHER GENES. Fourth messengers.")
10
MECHANISMS OF POSTSYNAPTIC ACTION OF PSYCHOACTIVE DRUGS
Altered synthesis of diverse types of proteins Acute changes in synaptic monoamines LONG-TERM ADAPTATIONS IN NEURONAL FUNCTION D phosphorylation of diverse types of phosphoproteins REGULATION OF NEURONAL GENE EXPRESSION R AC Dcyclic AMP G SHORT-TERM CHANGES IN NEURONAL FUNCTION STRESS: Gluco-corticoids GC receptor Nucleus Cell membrane Cytoplasm

11
Litio rappresenta lo stabilizzatore di prima scelta nella terapia e profilassi del disturbo bipolare negli studi di sospensione si è visto (14 studi) che il 50% dei pz in trattamento ricadeva entro 5 mesi dalla sospensione del litio, e che il rischio di ricaduta in una fase maniacale era 5 volte più frequente di quello di ricaduta in una fase depressiva vi sono dati recenti su un’efficacia specifica del litio sulla ideazione e mortalità suicidaria In una review della letteratura degli anni 90 (Goodwin e Jamison), basata su 10 studi in doppio cieco vs. placebo, la frequenza di ricadute in pz in trattamento con litio era significativamente minore di quelli in trattamento con placebo (34% vs. 81%) Fino all’80% dei pz bipolari trattati continuativamente con litio mostravano un miglioramento clinico In una review della letteratura degli anni 90 (Goodwin e Jamison), basata su 10 studi in doppio cieco vs. placebo, la frequenza di ricadute in pz in trattamento con litio era significativamente minore di quelli in trattamento con placebo (34% vs. 81%) Anche negli studi di sospensione si è visto (14 studi) che il 50% dei pz in trattamento ricadeva entro 5 mesi dalla sospensione del litio, e che il rischio di ricaduta in una fase maniacale era 5 volte più frequente di quello di ricaduta in una fase depressiva
 che il 50% dei pz in trattamento ricadeva entro 5 mesi dalla sospensione del litio, e che il rischio di ricaduta in una fase maniacale era 5 volte più frequente di quello di ricaduta in una fase depressiva. vi sono dati recenti su un’efficacia specifica del litio sulla ideazione e mortalità suicidaria. In una review della letteratura degli anni 90 (Goodwin e Jamison), basata su 10 studi in doppio cieco vs. placebo, la frequenza di ricadute in pz in trattamento con litio era significativamente minore di quelli in trattamento con placebo (34% vs. 81%) Fino all’80% dei pz bipolari trattati continuativamente con litio mostravano un miglioramento clinico. In una review della letteratura degli anni 90 (Goodwin e Jamison), basata su 10 studi in doppio cieco vs. placebo, la frequenza di ricadute in pz in trattamento con litio era significativamente minore di quelli in trattamento con placebo (34% vs. 81%) Anche negli studi di sospensione si è visto (14 studi) che il 50% dei pz in trattamento ricadeva entro 5 mesi dalla sospensione del litio, e che il rischio di ricaduta in una fase maniacale era 5 volte più frequente di quello di ricaduta in una fase depressiva.")
12
Litio Fattori predittivi di una risposta favorevole al litio:
sintomatologia maniacale senza aspetti disforici o gravemente psicotici ciclicità del disturbo con episodi non troppo ravvicinati nel tempo assenza di uso di sostanze pregressa risposta al litio

13
Litio - elettroliti - funzione tiroidea (T3, T4, TSH) - ECG
Esami da eseguire prima e durante il trattamento: - emocromo, azotemia e creatinina - elettroliti - funzione tiroidea (T3, T4, TSH) - ECG
 - ECG.")
14
Effetti collaterali del Litio
Disturbi neurologici: tremori fini, astenia muscolare, atassia, disturbi extrapiramidali (associato a neurolettici), riduzione della soglia convulsivante, aggravamento della miastenia gravis Disturbi cognitivi: rallentamento psichico, disturbi dell’attenzione, della memoria Disturbi gastrointestinali: nausea, vomito, diarrea Disturbi endocrini e metabolici: aumento ponderale, ipotiroidismo subclinico e clinico Sintomi renali: poliuria e polidipsia; diabete insipido; nefropatia del tubulo interstiziale (attualmente si tende ad escludere una tossicità glomerulare diretta del litio a dosaggi terapeutici) Disturbi cardiovascolari: appiattimento dell’onda T; allungamento del QRS Disturbi dermatologici: comparsa di alopecia areata, acne, aggravamento della psoriasi L’ipotiroidismo clinico ha una frequenza del 5% ed è più frequente nel genere femminile e in soggetti con un andamento rapido della ciclicità del disturbo Le modificazioni del peso corporeo hanno una frequenza di ca 1/3 dei pz e sembrano essere dose-dipendenti (litiemia > 0,8 mEq), più frequenti nel sesso femminile e in soggetti già in sovrappeso. Gli effetti cardiaci del litio, che dal punto di vista elettrocardiografico (ECG) sono simili a quelli dell'ipokaliemia, sono causati dallo spiazzamento del potassio intracellulare da parte del litio. Le modificazioni ECG più comuni sono appiattimento o l'inversione dell'onda T. Le modificazioni sono benigne e regrediscono dopo l'eliminazione del litio dall'organismo. Tuttavia, è essenziale eseguire un ECG prima dell'inizio della terapia e ripeterlo una volta all'anno. Il più comune effetto collaterale renale del litio è la poliuria con polidipsia secondaria. Il sintomo rappresenta un problema notevole per il 25-35% dei pazienti, che possono produrre oltre 3 litri di urine al giorno (range normale, 1-2 litri/die). La poliuria è causata da un antagonismo del litio nei confronti degli effetti dell'ormone antidiuretico, con una conseguente riduzione del riassorbimento di liquidi dal tubulo distale del rene. La poliuria potrebbe essere abbastanza significativa da rendere necessaria la valutazione della funzione renale del paziente - raccolta delle urine delle 24 ore per valutare la clearance della creatinina ed eventuale richiesta della consulenza di un nefrologo. Gli effetti collaterali renali più gravi, che si manifestano raramente, sono la glomerulonefrite, la nefrite interstiziale e l'insufficienza renale. Attualmente si ritiene che l'incidenza di queste complicazioni renali gravi sia maggiore di quanto si pensasse un tempo; pertanto, le si dovrebbero prendere in considerazione ogni qual volta siano suggerite dal quadro clinico
, riduzione della soglia convulsivante, aggravamento della miastenia gravis. Disturbi cognitivi: rallentamento psichico, disturbi dell’attenzione, della memoria. Disturbi gastrointestinali: nausea, vomito, diarrea. Disturbi endocrini e metabolici: aumento ponderale, ipotiroidismo subclinico e clinico. Sintomi renali: poliuria e polidipsia; diabete insipido; nefropatia del tubulo interstiziale (attualmente si tende ad escludere una tossicità glomerulare diretta del litio a dosaggi terapeutici) Disturbi cardiovascolari: appiattimento dell’onda T; allungamento del QRS. Disturbi dermatologici: comparsa di alopecia areata, acne, aggravamento della psoriasi. L’ipotiroidismo clinico ha una frequenza del 5% ed è più frequente nel genere femminile e in soggetti con un andamento rapido della ciclicità del disturbo. Le modificazioni del peso corporeo hanno una frequenza di ca 1/3 dei pz e sembrano essere dose-dipendenti (litiemia > 0,8 mEq), più frequenti nel sesso femminile e in soggetti già in sovrappeso. Gli effetti cardiaci del litio, che dal punto di vista elettrocardiografico (ECG) sono simili a quelli dell ipokaliemia, sono causati dallo spiazzamento del potassio intracellulare da parte del litio. Le modificazioni ECG più comuni sono appiattimento o l inversione dell onda T. Le modificazioni sono benigne e regrediscono dopo l eliminazione del litio dall organismo. Tuttavia, è essenziale eseguire un ECG prima dell inizio della terapia e ripeterlo una volta all anno. Il più comune effetto collaterale renale del litio è la poliuria con polidipsia secondaria. Il sintomo rappresenta un problema notevole per il 25-35% dei pazienti, che possono produrre oltre 3 litri di urine al giorno (range normale, 1-2 litri/die). La poliuria è causata da un antagonismo del litio nei confronti degli effetti dell ormone antidiuretico, con una conseguente riduzione del riassorbimento di liquidi dal tubulo distale del rene. La poliuria potrebbe essere abbastanza significativa da rendere necessaria la valutazione della funzione renale del paziente - raccolta delle urine delle 24 ore per valutare la clearance della creatinina ed eventuale richiesta della consulenza di un nefrologo. Gli effetti collaterali renali più gravi, che si manifestano raramente, sono la glomerulonefrite, la nefrite interstiziale e l insufficienza renale. Attualmente si ritiene che l incidenza di queste complicazioni renali gravi sia maggiore di quanto si pensasse un tempo; pertanto, le si dovrebbero prendere in considerazione ogni qual volta siano suggerite dal quadro clinico.")
15
Effetti avversi soggettivi e impatto sulla non compliance
% Importanza relativa sulla non-compliance Excessive thirst 35,9 … Polyuria 30,4 4 Memory disturbance 28,2 1 Tremor 26,9 3 Body weight 18,9 2 Somnolence/tiredness 12,4 5 Diarrhoea 8,7 Any disturbance 73,8 No disturbance 26,2 Dunner,

16
Sistema del GABA L’acido γ-aminobutirrico (GABA) rappresenta il principale sistema neurotrasmettitoriale inibitorio del SNC ed è uno dei neurotrasmettitori più rappresentati e presente in elevate concentrazioni (50% delle sinapsi inibitorie) Esistono tre sottotipi di recettori del GABA, A, B e C, e la maggior parte dei neuroni del SNC possiede almeno il sottotipo A, spesso anche il recettore B. Sono complessi macromolecolari localizzati a livello post-sinaptico e composti da diverse subunità e siti recettoriali (GABA, BDZ, etanolo, barbiturici, ecc.) Nel SNC dei mammiferi prevale il recettore GABA A Le BDZ agiscono legandosi alla sub-unità γ, potenziando l’azione GABAergica
 rappresenta il principale sistema neurotrasmettitoriale inibitorio del SNC ed è uno dei neurotrasmettitori più rappresentati e presente in elevate concentrazioni (50% delle sinapsi inibitorie) Esistono tre sottotipi di recettori del GABA, A, B e C, e la maggior parte dei neuroni del SNC possiede almeno il sottotipo A, spesso anche il recettore B. Sono complessi macromolecolari localizzati a livello post-sinaptico e composti da diverse subunità e siti recettoriali (GABA, BDZ, etanolo, barbiturici, ecc.) Nel SNC dei mammiferi prevale il recettore GABA A. Le BDZ agiscono legandosi alla sub-unità γ, potenziando l’azione GABAergica.")
17
Circuiti GABAergici nel cervello umano
GABA pathways in a normal brain GABA is the main inhibitory neurotransmitter in the central nervous system (CNS). GABAergic inhibition is seen at all levels of the CNS, including the hypothalamus, hippocampus, cerebral cortex and cerebellar cortex. As well as the large well-established GABA pathways, GABA interneurones are abundant in the brain, with 50% of the inhibitory synapses in the brain being GABA mediated. Basic and clinical pharmacology, 8th edition. Katzung BG. USA: The McGraw Hill Companies, Inc, 2001 Bloom FE. Neurohumoral transmission and the central nervous system. In: Goodman and Gilman’s the pharmacological basis of therapeutics, volume 1, 8th edition. Gilman et al. Singapore. McGraw-Hill Inc, 1992:244–268
. GABAergic inhibition is seen at all levels of the CNS, including the hypothalamus, hippocampus, cerebral cortex and cerebellar cortex. As well as the large well-established GABA pathways, GABA interneurones are abundant in the brain, with 50% of the inhibitory synapses in the brain being GABA mediated. Basic and clinical pharmacology, 8th edition. Katzung BG. USA: The McGraw Hill Companies, Inc, Bloom FE. Neurohumoral transmission and the central nervous system. In: Goodman and Gilman’s the pharmacological basis of therapeutics, volume 1, 8th edition. Gilman et al. Singapore. McGraw-Hill Inc, 1992:244–268.")
18
Il recettore GABAA The GABA A receptor
The GABA A receptor is a heteropentameric glycoprotein. A total of 5 distinct polypeptide subunits have been cloned to date; a, b, g, d and r, and multiple isoforms of these subunits are reported in the literature. Different confirmations of the GABA-A receptor are found throughout the brain, and the most common mammalian arrangements of sub-units is (a1)2(b2)2(g1). The specific subunits in the GABA-A receptor confer functional diversity on the receptor. For example, the g subunit needs to be co-expressed with the a and b sub-units to observe the potentiation of the GABA A receptor by benzodiazepines. Paul, SM. GABA and Glycine. The American College of Neuropsychopharmacology. Accessed on 16 September 2002
2(b2)2(g1). The specific subunits in the GABA-A receptor confer functional diversity on the receptor. For example, the g subunit needs to be co-expressed with the a and b sub-units to observe the potentiation of the GABA A receptor by benzodiazepines. Paul, SM. GABA and Glycine. The American College of Neuropsychopharmacology. Accessed on 16 September")
19
Il recettore GABAA The GABA A receptor
GABA is the major inhibitory neurotransmitter in the central nervous system. The GABA A receptor is composed of five sub-units – two alpha, two beta and one gamma sub-unit. Two molecules of GABA activate the receptor by binding to the alpha sub-units. Once activated the receptor allows the passage of negatively charged ions into the cytoplasm, which results in hyperpolarization and the inhibition of neurotransmission. Basic and clinical pharmacology, 8th edition. Katzung BG. USA: The McGraw Hill Companies, Inc, 2001

20
Sistema del Glutammato
Il glutammato è un neurotrasmettitore di natura aminoacidica. Viene sintetizzato a partire dalla glutamina proveniente dai neuroni glutammatargici e dalle cellule gliali circostanti ed è abbastanza diffuso in tutto il SNC La sua rimozione dalla sinapsi avviene attraverso due pompe di trasporto: una è un trasportatore pre-sinaptico, l’altra è localizzata nella glia circostante Il glutammato è uno dei due principali aminoacidi eccitatori del SNC e sembra svolgere un ruolo importante nella plasticità neuronale, nell’apprendimento e nella memoria Quando rimane in concentrazioni elevate nella sinapsi può avere effetti tossici, provocando morte cellulare, sia ritardata che immediata. Il ruolo principale del Glutammato nel SNC è più come aminoacido nella costituzione delle proteine che come neurotrasmettitore. Come l’aspartato è un potente aminoacido eccitatorio. Antagonisti dei recettori NMDA provocano sintomi simili alla schizofrenia nei soggetti sani, e aggravano la sintomatologia nei soggetti schizofrenici Numerosi dati clinici(35) e pre-clinici(36) convergono nell’ipotizzare un coinvolgimento del glutammato nella patogenesi della schizofrenia (ipotesi dell’ipofunzione dei recettori NMDA nella schizofrenia)(37). Antagonisti dei recettori NMDA (N-metil-D-Aspartato) producono effetti simili ai segni e sintomi della schizofrenia. Già si era visto che la PCP era in grado di produrre allucinazioni, delirio, disordini del pensiero, appiattimento emotivo, deficit cognitivi; in alcuni casi l’ingestione di PCP, rendeva il soggetto, in genere consumatore abituale, difficilmente distinguibile da pazienti schizofrenici(38). Rispetto alla PCP, la ketamina ha effetti meno intensi e prolungati, probabilmente a causa della minore affinità con cui si lega ai recettori NMDA(39) e della più breve emivita plasmatica. L’ipotesi della ketamina quale utile modello farmacologico di psicosi è sostenuta da diverse osservazioni(40,41). Studi effettuati su volontari sani(40,41,42), hanno evidenziato che la ketamina già a dosi subanestetiche produce sintomi positivi e negativi simili a quelli della schizofrenia, oltre ad alterazioni delle funzioni cognitive (diminuzione della attenzione, della memoria e della capacità di giudizio)(41,42,43). In realtà un recente studio(44), a conferma di precedenti osservazioni, ha evidenziato una relativa carenza di sintomi positivi (allucinazioni e delirio) in soggetti sani a cui veniva somministrata ketamina in dosi superiori a 0,1 mg/kg. Sembrerebbe pertanto che un’intossicazione acuta da ketamina si accompagni principalmente ad effetti riconducibili ai deficit cognitivi ed ai sintomi negativi della schizofrenia (mancanza di motivazioni e appiattimento affettivo). Ulteriore conferma della validità di tale modello deriva da uno studio di Curran e Monaghan(9) che osservarono l’insorgenza di specifici sintomi in pazienti schizofrenici stabili cronici dopo somministrazione di ketamina. L’importanza dell’ipotesi glutammatergica, rispetto ad altri modelli fisiopatologici della schizofrenia, quali l’ipotesi dopaminergica o serotoninergica(40) (in grado di spiegare soprattutto le forme paranoidee della psicosi e quindi le manifestazioni positive), risiede nella capacità di riprodurre con farmaci antagonisti dei recettori NDMA i sintomi negativi della schizofrenia. Lo spettro degli effetti prodotti dalla ketamina (in particolare i deficit cognitivi) in soggetti sani, riproduce quelli osservati nei pazienti con schizofrenia non-paranoidea. Tra l’altro la somministrazione di agonisti dopaminergici a schizofrenici può indurre un’eterogenea gamma di sintomi, anche se principalmente, come detto, di natura paranoidea e la risposta può essere quindi predittiva di ricadute della malattia in maniera meno sicura rispetto alla risposta indotta dalla ketamina(42). La psicosi da ketamina implicherebbe quindi una certa indipendenza dall’iperattivazione dei recettori dopaminergici(38). In effetti, la psicosi da ketamina non è migliorata dal trattamento con aloperidolo, uno dei principali antipsicotici(45). La patogenesi degli effetti psicotici della ketamina non è stata ancora definita, anche se da evidenze precliniche sembrerebbe che uno dei meccanismi principali risieda in una “disinibizione” del rilascio di glutammato(38,45). Antagonisti dei recettori NMDA bloccherebbero la stimolazione di neuroni GABA con conseguente mancata inibizione dei neuroni glutammatergici. Questa iperattivazione del glutammato sembrerebbe in grado di spiegare anche fenomeni di neurotossicità(46) e l’aumentato metabolismo cerebrale successivo alla somministrazione di ketamina(38). Ulteriori chiarimenti sull’effettivo ruolo del processo di iperattivazione glutammatergica, potrebbe avere importanti implicazioni sul trattamento della schizofrenia. Si è prospettato a tale scopo infatti la possibilità di ricorrere a farmaci che agiscono come inibitori del rilascio di glutammato quali la lamotrigina(43), che si è dimostrata attenuare gli effetti psicotici associati alla somministrazione di ketamina in soggetti sani. La Fenciclidina venne introdotta in terapia come anestetico dissociativo negli anni Cinquanta e poi abbandonata per l’alta incidenza di deliri ed allucinazioni post-operatori(2). Negli anni Settanta divenne farmaco di abuso, consumato principalmente in tre modi: inalato, fumato o ingerito. Quando viene fumato può essere mescolato anche con marijuana. Essendo sotto forma di polvere viene anche preparato in soluzione per uso iniettivo: gli effetti di induzione di uno stato confusionale, con possibile deliri e presenza di allucinazioni possono trapassare in una franca sintomatologia psicotica di tipo schizofrenico.
 e pre-clinici(36) convergono nell’ipotizzare un coinvolgimento del glutammato nella patogenesi della schizofrenia (ipotesi dell’ipofunzione dei recettori NMDA nella schizofrenia)(37). Antagonisti dei recettori NMDA (N-metil-D-Aspartato) producono effetti simili ai segni e sintomi della schizofrenia. Già si era visto che la PCP era in grado di produrre allucinazioni, delirio, disordini del pensiero, appiattimento emotivo, deficit cognitivi; in alcuni casi l’ingestione di PCP, rendeva il soggetto, in genere consumatore abituale, difficilmente distinguibile da pazienti schizofrenici(38). Rispetto alla PCP, la ketamina ha effetti meno intensi e prolungati, probabilmente a causa della minore affinità con cui si lega ai recettori NMDA(39) e della più breve emivita plasmatica. L’ipotesi della ketamina quale utile modello farmacologico di psicosi è sostenuta da diverse osservazioni(40,41). Studi effettuati su volontari sani(40,41,42), hanno evidenziato che la ketamina già a dosi subanestetiche produce sintomi positivi e negativi simili a quelli della schizofrenia, oltre ad alterazioni delle funzioni cognitive (diminuzione della attenzione, della memoria e della capacità di giudizio)(41,42,43). In realtà un recente studio(44), a conferma di precedenti osservazioni, ha evidenziato una relativa carenza di sintomi positivi (allucinazioni e delirio) in soggetti sani a cui veniva somministrata ketamina in dosi superiori a 0,1 mg/kg. Sembrerebbe pertanto che un’intossicazione acuta da ketamina si accompagni principalmente ad effetti riconducibili ai deficit cognitivi ed ai sintomi negativi della schizofrenia (mancanza di motivazioni e appiattimento affettivo). Ulteriore conferma della validità di tale modello deriva da uno studio di Curran e Monaghan(9) che osservarono l’insorgenza di specifici sintomi in pazienti schizofrenici stabili cronici dopo somministrazione di ketamina. L’importanza dell’ipotesi glutammatergica, rispetto ad altri modelli fisiopatologici della schizofrenia, quali l’ipotesi dopaminergica o serotoninergica(40) (in grado di spiegare soprattutto le forme paranoidee della psicosi e quindi le manifestazioni positive), risiede nella capacità di riprodurre con farmaci antagonisti dei recettori NDMA i sintomi negativi della schizofrenia. Lo spettro degli effetti prodotti dalla ketamina (in particolare i deficit cognitivi) in soggetti sani, riproduce quelli osservati nei pazienti con schizofrenia non-paranoidea. Tra l’altro la somministrazione di agonisti dopaminergici a schizofrenici può indurre un’eterogenea gamma di sintomi, anche se principalmente, come detto, di natura paranoidea e la risposta può essere quindi predittiva di ricadute della malattia in maniera meno sicura rispetto alla risposta indotta dalla ketamina(42). La psicosi da ketamina implicherebbe quindi una certa indipendenza dall’iperattivazione dei recettori dopaminergici(38). In effetti, la psicosi da ketamina non è migliorata dal trattamento con aloperidolo, uno dei principali antipsicotici(45). La patogenesi degli effetti psicotici della ketamina non è stata ancora definita, anche se da evidenze precliniche sembrerebbe che uno dei meccanismi principali risieda in una disinibizione del rilascio di glutammato(38,45). Antagonisti dei recettori NMDA bloccherebbero la stimolazione di neuroni GABA con conseguente mancata inibizione dei neuroni glutammatergici. Questa iperattivazione del glutammato sembrerebbe in grado di spiegare anche fenomeni di neurotossicità(46) e l’aumentato metabolismo cerebrale successivo alla somministrazione di ketamina(38). Ulteriori chiarimenti sull’effettivo ruolo del processo di iperattivazione glutammatergica, potrebbe avere importanti implicazioni sul trattamento della schizofrenia. Si è prospettato a tale scopo infatti la possibilità di ricorrere a farmaci che agiscono come inibitori del rilascio di glutammato quali la lamotrigina(43), che si è dimostrata attenuare gli effetti psicotici associati alla somministrazione di ketamina in soggetti sani. La Fenciclidina venne introdotta in terapia come anestetico dissociativo negli anni Cinquanta e poi abbandonata per l’alta incidenza di deliri ed allucinazioni post-operatori(2). Negli anni Settanta divenne farmaco di abuso, consumato principalmente in tre modi: inalato, fumato o ingerito. Quando viene fumato può essere mescolato anche con marijuana. Essendo sotto forma di polvere viene anche preparato in soluzione per uso iniettivo: gli effetti di induzione di uno stato confusionale, con possibile deliri e presenza di allucinazioni possono trapassare in una franca sintomatologia psicotica di tipo schizofrenico.")
21
Recettori del Glutammato
Recettori ionotropi : Recettori metabotropi NMDA AMPA Kainato

22
Recettore NMDA Il recettore NMDA è un recettore postsinaptico dell'acido glutammico. È un recettore ionotropico (una classe di recettori che funzionano essi stessi da canali ionici dopo legame col rispettivo ligando e/o attivazione da parte di altri fattori) che lascia fluire ioni Na+ e Ca2+ all'interno del neurone e ioni K+ al suo esterno. È costituito da svariate subunità, le più interne delle quali costituiscono la parete del canale ionotropico che permette il flusso degli ioni attraverso la membrana plasmatica Il recettore NMDA prende il nome da una molecola (N-Metil-D-Aspartato) che lo regola positivamente una volta legato al suo sito specifico. Infatti il recettore NMDA è un recettore ionotropico finemente regolabile che presenta vari siti di legame con i rispettivi composti
 che lascia fluire ioni Na+ e Ca2+ all interno del neurone e ioni K+ al suo esterno. È costituito da svariate subunità, le più interne delle quali costituiscono la parete del canale ionotropico che permette il flusso degli ioni attraverso la membrana plasmatica. Il recettore NMDA prende il nome da una molecola (N-Metil-D-Aspartato) che lo regola positivamente una volta legato al suo sito specifico. Infatti il recettore NMDA è un recettore ionotropico finemente regolabile che presenta vari siti di legame con i rispettivi composti.")
23
Ipotesi glutammatergica della Schizofrenia
Antagonisti dei recettori NMDA (PCP, ketamina) provocano sintomi simili alla schizofrenia nei soggetti sani (sintomi psicotici, ritiro sociale e deficit di working memory), e aggravano la sintomatologia nei soggetti schizofrenici uno dei meccanismi principali sembra risiedere in una riduzione del controllo sul sistema GABAergico, mediato dal recettore NMDA, con “disinibizione” del rilascio di glutammato La sregolazione glutammatergica è alla base di fenomeni di neurotossicità L’ipotesi iniziale di una ipofunzione glutamatergica negli schizofrenici fu fortemente influenzata da studi di neuroimaging che suggerivano una ipofunzione corticale a riposo e durante tasks cognitivi. In seguito invece si evidenziò che gli antagonisti del recettore NMDA preferenzialmente incrementavano il metabolismo e i livelli di glutammato extracellulare in circuiti limbici definiti (Duncan e al, 1999; Holcomb e al, 2005). Questi studi suggerivano che dosi subanestetiche di antagonisti del recettore NMDA erano in grado di attivare il recettore NMDA nella corteccia cerebrale e nell’ippocampo; il glutammato rilasciato da questi neuroni può attivare altri recettori ionotropi del glutammato (es kainato) o rettori metabotropici. E’ probabile che l’azione dei bloccanti il recettore NMDA (iperattivazione glutammatergica) avvenga attraverso gli interneuroni GABAergici che espongono recettori NMDA (questi svolgono un’azione di controllo sul sistema GABA che a sua volta ha un’azione inibitoria regolatoria sul sistema gluatamatergico eccitatorio). Da questo punto di vista è più di 10 anni che la ricerca sta testando farmaci agonisti dei recettori matabotropici mGlu2/3 per trattare sintomi positivi e negativi dei pz con schizofrenia. Uno dei più riconosciuti effetti farmaacologici degli agonisti mGlu2/3 è l’annullamento di vari effetti indotti da sostanze psicotomimetiche come la PCP o la Ketamina. Il rilascio di Glutammato corticale è anche sotto il controllo dei recettori 5-HT2a. L’attivazione dei 5-HT2a induce correnti eccitatorie post-sinaptiche a livello dei neuroni piramidali in corteccia pre-frontale. L’incremento del rilascio di Glutammato mediato dai recettori 5-HT2a si è visto legato a distorsioni cognitive, percettive e affettive prodotte da allucinogeni agonisti 5-HT2a (come l’LSD). E’ interessante considerare che questi effetti possono essere bloccati non solo dagli antagonisti 5HT2a ma anche dagli agonisti del recettore mGlu2/3 (Marek e al, 2000). Tale interazione tra 5-HT2a e mGlu2/3 è anche supportata da evidenze emergenti di una colocalizzazione di 5-HT2a e mGlu2/3 in corteccia (Gonzales-Maeso e al, 2008). Uno dei più documentati effetti comportamentali della stimolazione dei recettori mGlu2/3 è l’annullamento dei comportamenti indotti da da agenti PCP-like e 5-HT2a agonisti. E’ probabile che l’aumento della trasmissione glutamatergica in corteccia prefrontale determini una attività neuronale desincronizzata che può essere espressa funzionalmente da una condizione di ipofrontalità (ad es. prestazioni deficitarie ai working memory tasks) (Sebban e al, 2002; Pinault, 2008). Esiste pertanto una relazione tra eccessivo rilascio di glutammato, perdita della sincronizzazione neuronale e ipofrontalità indotte da sostanze psicotomimetiche e/o osservate in pz con schizofrenia. La stimolazione dei recettori mGlu2/3 può annullare l’eccessivo rilascio di glutammato, ripristinare la normale attività neuronale e quindi migliorare le performance cognitive (Moghaddam e Adams, 1998; Krystal e al, 2005). Tra i più studiati, l’agonista mGlu2/3 LY si è visto ridurre in maniera significativa i sintomi positivi e negativi dei pz con schizofrenia (Patil e al, 2007).
 provocano sintomi simili alla schizofrenia nei soggetti sani (sintomi psicotici, ritiro sociale e deficit di working memory), e aggravano la sintomatologia nei soggetti schizofrenici. uno dei meccanismi principali sembra risiedere in una riduzione del controllo sul sistema GABAergico, mediato dal recettore NMDA, con disinibizione del rilascio di glutammato. La sregolazione glutammatergica è alla base di fenomeni di neurotossicità. L’ipotesi iniziale di una ipofunzione glutamatergica negli schizofrenici fu fortemente influenzata da studi di neuroimaging che suggerivano una ipofunzione corticale a riposo e durante tasks cognitivi. In seguito invece si evidenziò che gli antagonisti del recettore NMDA preferenzialmente incrementavano il metabolismo e i livelli di glutammato extracellulare in circuiti limbici definiti (Duncan e al, 1999; Holcomb e al, 2005). Questi studi suggerivano che dosi subanestetiche di antagonisti del recettore NMDA erano in grado di attivare il recettore NMDA nella corteccia cerebrale e nell’ippocampo; il glutammato rilasciato da questi neuroni può attivare altri recettori ionotropi del glutammato (es kainato) o rettori metabotropici. E’ probabile che l’azione dei bloccanti il recettore NMDA (iperattivazione glutammatergica) avvenga attraverso gli interneuroni GABAergici che espongono recettori NMDA (questi svolgono un’azione di controllo sul sistema GABA che a sua volta ha un’azione inibitoria regolatoria sul sistema gluatamatergico eccitatorio). Da questo punto di vista è più di 10 anni che la ricerca sta testando farmaci agonisti dei recettori matabotropici mGlu2/3 per trattare sintomi positivi e negativi dei pz con schizofrenia. Uno dei più riconosciuti effetti farmaacologici degli agonisti mGlu2/3 è l’annullamento di vari effetti indotti da sostanze psicotomimetiche come la PCP o la Ketamina. Il rilascio di Glutammato corticale è anche sotto il controllo dei recettori 5-HT2a. L’attivazione dei 5-HT2a induce correnti eccitatorie post-sinaptiche a livello dei neuroni piramidali in corteccia pre-frontale. L’incremento del rilascio di Glutammato mediato dai recettori 5-HT2a si è visto legato a distorsioni cognitive, percettive e affettive prodotte da allucinogeni agonisti 5-HT2a (come l’LSD). E’ interessante considerare che questi effetti possono essere bloccati non solo dagli antagonisti 5HT2a ma anche dagli agonisti del recettore mGlu2/3 (Marek e al, 2000). Tale interazione tra 5-HT2a e mGlu2/3 è anche supportata da evidenze emergenti di una colocalizzazione di 5-HT2a e mGlu2/3 in corteccia (Gonzales-Maeso e al, 2008). Uno dei più documentati effetti comportamentali della stimolazione dei recettori mGlu2/3 è l’annullamento dei comportamenti indotti da da agenti PCP-like e 5-HT2a agonisti. E’ probabile che l’aumento della trasmissione glutamatergica in corteccia prefrontale determini una attività neuronale desincronizzata che può essere espressa funzionalmente da una condizione di ipofrontalità (ad es. prestazioni deficitarie ai working memory tasks) (Sebban e al, 2002; Pinault, 2008). Esiste pertanto una relazione tra eccessivo rilascio di glutammato, perdita della sincronizzazione neuronale e ipofrontalità indotte da sostanze psicotomimetiche e/o osservate in pz con schizofrenia. La stimolazione dei recettori mGlu2/3 può annullare l’eccessivo rilascio di glutammato, ripristinare la normale attività neuronale e quindi migliorare le performance cognitive (Moghaddam e Adams, 1998; Krystal e al, 2005). Tra i più studiati, l’agonista mGlu2/3 LY si è visto ridurre in maniera significativa i sintomi positivi e negativi dei pz con schizofrenia (Patil e al, 2007).")
24
Ipotesi glutammatergica della Schizofrenia
Uno dei più documentati effetti comportamentali della stimolazione dei recettori mGlu2/3 è l’annullamento dei comportamenti indotti da da agenti PCP-like e 5-HT2a agonisti E’ probabile che l’aumento della trasmissione glutamatergica in corteccia prefrontale determini una attività neuronale desincronizzata che può essere espressa funzionalmente da una condizione di ipofrontalità (ad es. prestazioni deficitarie ai working memory tasks)
")
25
Ipotesi glutammatergica della Schizofrenia
La stimolazione dei recettori mGlu2/3 può annullare l’eccessivo rilascio di glutammato, ripristinare la normale attività neuronale e quindi migliorare la sintomatologia negativa della schizofrenia e le funzioni di working memory (LY )
")
26
Circuiti glutamatergici nel cervello umano
The glutamate pathways in the ‘normal’ brain In the normal brain the prominent glutamatergic pathways are: the cortico-cortical pathways; the pathways between the thalamus and the cortex; and the extrapyramidal pathway (the projections between the cortex and striatum). Other glutamate projections exist between the cortex, substantia nigra, subthalmic nucleus and pallidum. Glutamate-containing neuronal terminals are ubiquitous in the central nervous system and their importance in mental activity and neurotransmission is considerable. Int Clin Psychopharmacol 1995;10(Suppl 3):21–28 Brain structures and neuronal projections. Institute of Chemistry, Pharmacology and Biomedical Sciences. University of Sunderland. Accessed on 21 March 2003
. Other glutamate projections exist between the cortex, substantia nigra, subthalmic nucleus and pallidum. Glutamate-containing neuronal terminals are ubiquitous in the central nervous system and their importance in mental activity and neurotransmission is considerable. Int Clin Psychopharmacol 1995;10(Suppl 3):21–28. Brain structures and neuronal projections. Institute of Chemistry, Pharmacology and Biomedical Sciences. University of Sunderland. Accessed on 21 March")
27
Meccanismi d’azione degli stabilizzatori
agiscono sui canali del Na e del Ca voltaggio-dipendenti aumentando la stabilità di membrana potenziano l’attività GABAergica inibiscono la trasmissione glutammatergica

28
K+ 20:1 K+ Il rapporto scritto sopra la freccia indica quanto maggiormente è concentrato uno ione da un lato rispetto all’altro Na+ 1:10 Na+ La freccia indica la direzione in cui gli ioni sono sospinti dal loro gradiente di concentrazione Cl- 1:11 Cl- 1:10.000 Ca++ Ca++ Ci sono 4 ioni importanti, K+, Na+, Ca++, Cl-. Tre sono più concentrati all’esterno, uno (K+) all’interno
 all’interno.")
29
Canali ionici e pompe di trasporto
Nella membrana cellulare esistono delle proteine specializzate, chiamate canali del Sodio, del Calcio, del Potassio attraverso i quali passano gli ioni; ed esistono delle pompe del Sodio e del Calcio che lavorano contro gradiente, per la differenza di concentrazioni degli ioni attraverso le membrane consumando energia

30
Canali ionici e pompe di trasporto
La pompa Sodio/Potassio facilita i movimenti di questi ioni attraverso la membrana , essa è di grande importanza perché mantiene alte le concentrazioni di Potassio e basse quelle di Sodio dentro la cellula

31
Il potassio, K+ è uno ione positivo (catione)
Il potassio, K+ è uno ione positivo (catione). Dato che è molto più concentrato all’interno* tenderà ad uscire rendendo l’interno della cellula ancor più negativo Anche il sodio, Na+ è uno ione positivo (catione). Dato che è molto più concentrato all’esterno tenderà ad entrare rendendo l’interno della cellula meno negativo Nella membrana cellulare esistono delle proteine specializzate, chiamate canali del Sodio, del calcio, del Potassio attraverso i quali passano gli ioni ed esistono delle pompe del Sodio e del calcio che lavorano contro gradiente per la differenza di concentrazioni degli ioni attraverso le membrane, consumando energia; è chiaro che tanto più aumentano le concentrazioni del Sodio e del calcio nel sangue tanto più aumenta il consumo di energia. La pompa Sodio/Potassio facilita i movimenti di questi ioni attraverso la membrana , essa è di grande importanza perché mantiene alte le concentrazioni di Potassio e basse quelle di Sodio dentro la cellula. Alte concentrazioni di Potassio dentro la cellula sono necessarie per numerosi processi : uno è la sintesi proteica nei ribosomi, un altro è che numerosi enzimi della glicolisi richiedono Potassio, per esempio la piruvato chinasi. La pompa del Sodio induce lo scambio di 3 ioni Sodio che escono dalla cellula contro 2 ioni di Potassio che entrano nella cellula; essa è costituita da 1318 aminoacidi ed è disposta lungo la membrana cellulare; anche la pompa del calcio ( composta da una singola proteina di 1220 aminoacidi ) estrude ioni calcio dalla cellula contro gradiente di concentrazione, consumando energia. E' stato calcolato che le cellule renali e del cervello usano il 70% della loro energia allo scopo di pompare Sodio fuori dalla cellula e Potassio dentro la cellula, nei globuli rossi per ogni scambio 3 ioni Sodio fuori 2 ioni Potassio dentro, viene usata una molecola di ATP. Le variazioni di permeabilità della membrana a questi due ioni sono in grado di determinare grandi cambiamenti nel potenziale di membrana
. Dato che è molto più concentrato all’interno* tenderà ad uscire rendendo l’interno della cellula ancor più negativo. Anche il sodio, Na+ è uno ione positivo (catione). Dato che è molto più concentrato all’esterno tenderà ad entrare rendendo l’interno della cellula meno negativo. Nella membrana cellulare esistono delle proteine specializzate, chiamate canali del Sodio, del calcio, del Potassio attraverso i quali passano gli ioni ed esistono delle pompe del Sodio e del calcio che lavorano contro gradiente per la differenza di concentrazioni degli ioni attraverso le membrane, consumando energia; è chiaro che tanto più aumentano le concentrazioni del Sodio e del calcio nel sangue tanto più aumenta il consumo di energia. La pompa Sodio/Potassio facilita i movimenti di questi ioni attraverso la membrana , essa è di grande importanza perché mantiene alte le concentrazioni di Potassio e basse quelle di Sodio dentro la cellula. Alte concentrazioni di Potassio dentro la cellula sono necessarie per numerosi processi : uno è la sintesi proteica nei ribosomi, un altro è che numerosi enzimi della glicolisi richiedono Potassio, per esempio la piruvato chinasi. La pompa del Sodio induce lo scambio di 3 ioni Sodio che escono dalla cellula contro 2 ioni di Potassio che entrano nella cellula; essa è costituita da 1318 aminoacidi ed è disposta lungo la membrana cellulare; anche la pompa del calcio ( composta da una singola proteina di 1220 aminoacidi ) estrude ioni calcio dalla cellula contro gradiente di concentrazione, consumando energia. E stato calcolato che le cellule renali e del cervello usano il 70% della loro energia allo scopo di pompare Sodio fuori dalla cellula e Potassio dentro la cellula, nei globuli rossi per ogni scambio 3 ioni Sodio fuori 2 ioni Potassio dentro, viene usata una molecola di ATP. Le variazioni di permeabilità della membrana a questi due ioni sono in grado di determinare grandi cambiamenti nel potenziale di membrana.")
32
Ad ogni ciclo, la pompa sodio-potassio - espelle tre ioni Na+ (Sodio)
La differenza di concentrazione degli ioni tra interno ed esterno della cellula è mantenuto dal lavoro incessante della pompa sodio-potassio (e da quello delle altre pompe, pompa del calcio, pompa del cloro) Ad ogni ciclo, la pompa sodio-potassio - espelle tre ioni Na+ (Sodio) - trasporta all’interno due ioni K+ (Potassio) consumando una molecola di ATP Nella membrana cellulare esistono delle proteine specializzate, chiamate canali del Sodio, del calcio, del Potassio attraverso i quali passano gli ioni ed esistono delle pompe del Sodio e del calcio che lavorano contro gradiente per la differenza di concentrazioni degli ioni attraverso le membrane, consumando energia; è chiaro che tanto più aumentano le concentrazioni del Sodio e del calcio nel sangue tanto più aumenta il consumo di energia. La pompa Sodio/Potassio facilita i movimenti di questi ioni attraverso la membrana , essa è di grande importanza perché mantiene alte le concentrazioni di Potassio e basse quelle di Sodio dentro la cellula. Alte concentrazioni di Potassio dentro la cellula sono necessarie per numerosi processi : uno è la sintesi proteica nei ribosomi, un altro è che numerosi enzimi della glicolisi richiedono Potassio, per esempio la piruvato chinasi. La pompa del Sodio induce lo scambio di 3 ioni Sodio che escono dalla cellula contro 2 ioni di Potassio che entrano nella cellula; essa è costituita da 1318 aminoacidi ed è disposta lungo la membrana cellulare; anche la pompa del calcio ( composta da una singola proteina di 1220 aminoacidi ) estrude ioni calcio dalla cellula contro gradiente di concentrazione, consumando energia. E' stato calcolato che le cellule renali e del cervello usano il 70% della loro energia allo scopo di pompare Sodio fuori dalla cellula e Potassio dentro la cellula, nei globuli rossi per ogni scambio 3 ioni Sodio fuori 2 ioni Potassio dentro, viene usata una molecola di ATP. L’80% dell’ATP consumato dal neurone è dovuto al funzionamento delle pompe ioniche
 Ad ogni ciclo, la pompa sodio-potassio. - espelle tre ioni Na+ (Sodio) - trasporta all’interno due ioni K+ (Potassio) consumando una molecola di ATP. Nella membrana cellulare esistono delle proteine specializzate, chiamate canali del Sodio, del calcio, del Potassio attraverso i quali passano gli ioni ed esistono delle pompe del Sodio e del calcio che lavorano contro gradiente per la differenza di concentrazioni degli ioni attraverso le membrane, consumando energia; è chiaro che tanto più aumentano le concentrazioni del Sodio e del calcio nel sangue tanto più aumenta il consumo di energia. La pompa Sodio/Potassio facilita i movimenti di questi ioni attraverso la membrana , essa è di grande importanza perché mantiene alte le concentrazioni di Potassio e basse quelle di Sodio dentro la cellula. Alte concentrazioni di Potassio dentro la cellula sono necessarie per numerosi processi : uno è la sintesi proteica nei ribosomi, un altro è che numerosi enzimi della glicolisi richiedono Potassio, per esempio la piruvato chinasi. La pompa del Sodio induce lo scambio di 3 ioni Sodio che escono dalla cellula contro 2 ioni di Potassio che entrano nella cellula; essa è costituita da 1318 aminoacidi ed è disposta lungo la membrana cellulare; anche la pompa del calcio ( composta da una singola proteina di 1220 aminoacidi ) estrude ioni calcio dalla cellula contro gradiente di concentrazione, consumando energia. E stato calcolato che le cellule renali e del cervello usano il 70% della loro energia allo scopo di pompare Sodio fuori dalla cellula e Potassio dentro la cellula, nei globuli rossi per ogni scambio 3 ioni Sodio fuori 2 ioni Potassio dentro, viene usata una molecola di ATP. L’80% dell’ATP consumato dal neurone è dovuto al funzionamento delle pompe ioniche.")
33
Carbamazepina Ha attualmente l’indicazione per la ‘mania’
stabilizza i canali del Na voltaggio dipendenti potenzia l’attività GABAergica a livello limbico (ippocampo); ↓ il rilascio di glutammato; ↓ il turn-over della DA è un potente induttore del citocromo P450 (riduzione dei livelli plasmatici di neurolettici e antidepressivi, fino all’80% del loro dosaggio)
; ↓ il rilascio di glutammato; ↓ il turn-over della DA. è un potente induttore del citocromo P450 (riduzione dei livelli plasmatici di neurolettici e antidepressivi, fino all’80% del loro dosaggio)")
34
Carbamazepina Effetti collaterali (più frequenti):
Neurologici: vertigini, cefalea, sonnolenza, diplopia, visione offuscata, atassia Intestinali: nausea, vomito, diarrea ma anche stipsi

35
Carbamazepina Effetti collaterali (meno frequenti)
leucopenia e piastrinopenia transitorie ↑ asintomatico degli enzimi epatici rush cutaneo di aspetto morbilliforme e prurito (2-12%) alterazioni della conduzione cardiaca, bradicardia reazioni idiosincrasiche: anemia aplastica e agranulocitosi (1,5 e 5 casi su un milione); sindr. di Stevens-Johnson (mortalità > 10%) Un numero di leucociti < 3000 mm3 e di neutrofili < 1500 mm3 richiede la sospensione del farmaco La sindrome di Stevens-Johnson è la forma estrema per gravità dell’eritema multiforme o eritema polimorfo, una caratteristica sindrome acuta da ipersensibilità, che può essere causata da varie cause: malattie virali, infezioni batteriche, allergia a farmaci, vaccinazioni ed altre ancora. Le lesioni classiche dell’eritema multiforme sono costituite da papule o chiazze arrossate, rotonde, con un diametro di 1-5 cm, con aspetto a bersaglio o a coccarda (cioè con una parte centrale di aspetto cianotico, a volte bolloso circondata da anelli concentrici di colore più o meno rosso). Nella sindrome di Stevens-Johnson le lesioni non si limitano alla cute, ma vengono coinvolte le mucose, particolarmente quelle della bocca (con frequente degenerazione crostosa della mucosa delle labbra), delle congiuntive e dell’uretra, e compaiono aree di necrosi e sfaldamento cutaneo. Lo stato generale è di regola compromesso con febbre elevata, tosse, mal di gola, vomito, diarrea, dolori articolari. Le lesioni cutaneo-mucose sono ingravescenti, con formazione estesa di bolle, spesso emorragiche; la perdita della barriera epidermica porta ad uno sbilancio di liquidi e sali minerali, con un alto rischio di infezioni batteriche secondarie. Ecco spiegato il motivo per cui qualche volta i pazienti con tale sindrome possono, nelle forme più gravi, richiedere il ricovero in un centro di terapia intensiva per grandi ustionati, e la ragione per cui il tasso di mortalità della malattia varia tra il 5 ed il 25%.Nella maggioranza dei casi comunque le lesioni cutaneo-mucose e la compromissione dello stato generale durano per 7-10 giorni, e la malattia si risolve completamente in un tempo variante tra 1 e 6 settimane.
 alterazioni della conduzione cardiaca, bradicardia. reazioni idiosincrasiche: anemia aplastica e agranulocitosi (1,5 e 5 casi su un milione); sindr. di Stevens-Johnson (mortalità > 10%) Un numero di leucociti < 3000 mm3 e di neutrofili < 1500 mm3 richiede la sospensione del farmaco. La sindrome di Stevens-Johnson è la forma estrema per gravità dell’eritema multiforme o eritema polimorfo, una caratteristica sindrome acuta da ipersensibilità, che può essere causata da varie cause: malattie virali, infezioni batteriche, allergia a farmaci, vaccinazioni ed altre ancora. Le lesioni classiche dell’eritema multiforme sono costituite da papule o chiazze arrossate, rotonde, con un diametro di 1-5 cm, con aspetto a bersaglio o a coccarda (cioè con una parte centrale di aspetto cianotico, a volte bolloso circondata da anelli concentrici di colore più o meno rosso). Nella sindrome di Stevens-Johnson le lesioni non si limitano alla cute, ma vengono coinvolte le mucose, particolarmente quelle della bocca (con frequente degenerazione crostosa della mucosa delle labbra), delle congiuntive e dell’uretra, e compaiono aree di necrosi e sfaldamento cutaneo. Lo stato generale è di regola compromesso con febbre elevata, tosse, mal di gola, vomito, diarrea, dolori articolari. Le lesioni cutaneo-mucose sono ingravescenti, con formazione estesa di bolle, spesso emorragiche; la perdita della barriera epidermica porta ad uno sbilancio di liquidi e sali minerali, con un alto rischio di infezioni batteriche secondarie. Ecco spiegato il motivo per cui qualche volta i pazienti con tale sindrome possono, nelle forme più gravi, richiedere il ricovero in un centro di terapia intensiva per grandi ustionati, e la ragione per cui il tasso di mortalità della malattia varia tra il 5 ed il 25%.Nella maggioranza dei casi comunque le lesioni cutaneo-mucose e la compromissione dello stato generale durano per 7-10 giorni, e la malattia si risolve completamente in un tempo variante tra 1 e 6 settimane.")
36
Carbamazepina Dosaggi: tra 200 e 1600mg (concentrazioni seriche tra 4 e 12 mg per ml) Esami da eseguire prima e durante il trattamento: emocromo con formula, got, gpt, γgt, bilirubina, fosfatasi alcalina; azotemia, elettroliti serici; Ecg per persone di età superiore a 40anni(da ripetere prima del trattamento e periodicamente durante il trattamento)
")
37
Valproato stabilizza i canali ionici voltaggio-dipendenti di K, Na e Ca ↑ il metabolismo del GABA, il rilascio del GABA dai terminali sinaptici, potenzia la trasmissione sinaptica GABAergica soprattutto a livello limbico- ippocampale attualmente ha indicazione per la cura e la prevenzione della mania La maggior parte dei dati si riferiscono al trattamento della mania in acuto, invece la sua efficacia nel mantenimento non è stata ancora sufficientemente dimostrata

38
Valproato ↑ transitorio delle transaminasi, fastidi gastrointestinali
trombocitopenia transitoria (in genere benigna e dose- dipendente) e allungamento del tempo di sanguinamento di scarsa rilevanza clinica (controlli periodici della crasi ematica, interazioni con farmaci inibitori della aggregazione piastrinica) ↑ di peso e caduta e variazione del colore dei capelli (fino al 70% e 43%) Iperammoniemia (encefalopatia iperammonemica) reazioni idiosincrasiche: pancreatite emorragica, insufficienza epatica irreversibile Per la pancreatite emorragica i fattori di rischio sono l’età inferiore ai 20 anni e una politerapia la contemporanea somministrazione di valproato e topiramato è stata associata all’insorgenza di encefalopatia e/o iperammoniemia
 e allungamento del tempo di sanguinamento di scarsa rilevanza clinica (controlli periodici della crasi ematica, interazioni con farmaci inibitori della aggregazione piastrinica) ↑ di peso e caduta e variazione del colore dei capelli (fino al 70% e 43%) Iperammoniemia (encefalopatia iperammonemica) reazioni idiosincrasiche: pancreatite emorragica, insufficienza epatica irreversibile. Per la pancreatite emorragica i fattori di rischio sono l’età inferiore ai 20 anni e una politerapia. la contemporanea somministrazione di valproato e topiramato è stata associata all’insorgenza di encefalopatia e/o iperammoniemia.")
39
Valproato Dosaggi: si può iniziare con 250mg tre volte/die e si prosegue aumentando di 250/500mg ogni 2/5 gg con dosaggi medi di mg (concentrazioni plasmatiche tra 45 e 100 mg/ml) (per pz ambulatoriali) Esami da eseguire prima e durante il trattamento: emocromo con formula, elettroliti serici, transaminasi, amilasi, (lipasi), bilirubinemia, pseudocolinesterasi, (da ripetere dopo il primo mese, poi ogni 6 mesi); PT, PTT, fibrinogeno (da ripetere ogni 6 mesi)
 (per pz ambulatoriali) Esami da eseguire prima e durante il trattamento: emocromo con formula, elettroliti serici, transaminasi, amilasi, (lipasi), bilirubinemia, pseudocolinesterasi, (da ripetere dopo il primo mese, poi ogni 6 mesi); PT, PTT, fibrinogeno (da ripetere ogni 6 mesi)")
40
Stabilizzatori di II generazione
Gabapentin ( mg/die) Pregabalin ( mg/die) Lamotrigina ( mg/die) Topiramato ( mg/die) Oxcarbazepina ( mg/die) Lamotrigina: ha l’indicazione per la prevenzione degli episodi depressivi nei pz con disturbo bipolare di tipo I che presentano prevalentemente episodi depressivi. Il farmaco non è indicato per il trattamento acuto degli episodi maniacali e depressivi. Gli effetti collaterali tipo reazioni cutanee (a volte anche gravi) hanno maggiori probabilità di comparire se la dose iniziale è troppo alta, se viene scalata troppo rapidamente o durante la concomitante assunzione di ac. Valproico. Nella maggior parte dei casi compaiono dopo 2-8 settimane di terapia. Le concentrazioni plasmatiche di lamotrigina vengono quasi raddoppiate dalla concomitante somministrazione di valproato. Anche la sertralina può aumentare la concentrazione plasmatica di lamotrigina (meno che il valproato). La carbamazepina può invece ridurre del 40/50% le concentrazioni plasmatiche di lamotrigina. Topiramato: monitorare i livelli serici di bicarbonato a causa del rischio di ac. metabolica ipercloremica. Oxcarbamazepina: controllare la sodiemia per la possibile iponatriemia soprattutto i primi tre mesi.
 Pregabalin ( mg/die) Lamotrigina ( mg/die) Topiramato ( mg/die) Oxcarbazepina ( mg/die) Lamotrigina: ha l’indicazione per la prevenzione degli episodi depressivi nei pz con disturbo bipolare di tipo I che presentano prevalentemente episodi depressivi. Il farmaco non è indicato per il trattamento acuto degli episodi maniacali e depressivi. Gli effetti collaterali tipo reazioni cutanee (a volte anche gravi) hanno maggiori probabilità di comparire se la dose iniziale è troppo alta, se viene scalata troppo rapidamente o durante la concomitante assunzione di ac. Valproico. Nella maggior parte dei casi compaiono dopo 2-8 settimane di terapia. Le concentrazioni plasmatiche di lamotrigina vengono quasi raddoppiate dalla concomitante somministrazione di valproato. Anche la sertralina può aumentare la concentrazione plasmatica di lamotrigina (meno che il valproato). La carbamazepina può invece ridurre del 40/50% le concentrazioni plasmatiche di lamotrigina. Topiramato: monitorare i livelli serici di bicarbonato a causa del rischio di ac. metabolica ipercloremica. Oxcarbamazepina: controllare la sodiemia per la possibile iponatriemia soprattutto i primi tre mesi.")
41
Effetti avversi degli anti-epilettici
Cogni-tive Hepatic Cuta-neous Haemato-logical body weight Fetal Valproate + ++ +++ Carbamazepine Lamotrigine - ? Gabapentin Topiramate Swann, 2001

Presentazioni simili