Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Riparazione del DNA, Checkpoints & Ciclo Cellulare

2
Fattori in grado di indurre lesioni nel DNA
Radiazioni ultraviolette Radiazioni ionizzanti Composti chimici naturali o di sintesi Specie reattive dell’ossigeno species La maggior parte di queste deriva da processi endogeni In un organismo umano ogni giorno il DNA subisce ~100,000,000,000,000,000 lesioni Mammalian cells are constantly at risk of damage to their DNA from multiple causes1, including ultraviolet light, ionizing radiation, reactive oxygen species, several endogenous and synthetic compounds, and during normal DNA replication. Approximately 1x104 DNA lesions (single-strand breaks [SSBs]) are generated in a metabolically active mammalian cell each day.1,2 There are approximately 1x1013 cells in the human body, so this corresponds to a total of around 1x1017 DNA lesions per person, per day.1-3 References 1. Jackson SP. Biochem Soc Trans 2001; 29: 2. Lindahl T. Nature 1993; 362: 3. Jackson SP, Bishop CL. Drug Discovery World 2003; Fall:
 are generated in a metabolically active mammalian cell each day.1,2 There are approximately 1x1013 cells in the human body, so this corresponds to a total of around 1x1017 DNA lesions per person, per day.1-3. References. 1. Jackson SP. Biochem Soc Trans 2001; 29: Lindahl T. Nature 1993; 362: Jackson SP, Bishop CL. Drug Discovery World 2003; Fall:")
3
Le cellule tumorali sono altamente suscettibili all’inibizione della riparazione del DNA
Proliferano in modo non regolato meno tempo a disposizione per riparare il DNA rispetto alle cellule normali Crescono in condizioni di stress, il che causa un danno continuo al DNA Presentano difetti nella riparazione del DNA fenotipo mutator La crescita continua nonostante l’instabilità genomica Sono dipendenti dalle vie di riparazione del DNA che ancora possiedono Eukaryotic cells have evolved mechanisms to monitor the integrity of their genome and repair damaged DNA before the cell cycle progresses. The mechanisms that monitor for damaged DNA are intimately linked with cell-cycle events and the process known as “checkpoint” control. Loss of checkpoint function predisposes a cell to acquire selective oncogenic mutations and, therefore, it is an important prognostic indicator. People with genetic instability disorders that cause defects in checkpoint function have an increased incidence of many cancers.1 Cancer cells undergo deregulated proliferation, losing cell-cycle control and resting in the G0 phase of the cell cycle for little or no time. Thus, there is less opportunity for DNA repair to occur than in normal cells. Growth under stress increases the risk of DNA damage in cancer cells. In addition, the genomic instability associated with cancer cell formation (“mutator phenotype”) results in an increased DNA mutation rate.2 Many DNA repair pathways are lost or deficient in cancer cells. In particular, breast or ovarian tumor cells with hereditary breast cancer associated gene (BRCA) 1 or BRCA2 mutations are HR deficient and so they lack the ability to efficiently repair DSBs.3 Cancer cells continue to grow despite ongoing genomic instability and so the repair pathways they still retain become integral to their survival. Interest in targeting these remaining DNA repair pathways in cancer cells as a selective anti-cancer therapy is developing. References 1. Khanna K. J Natl Cancer Inst 2000; 92: 2. Bielas JH, Loeb LA. Environ Mol Mutagen 2005; 45: 3. Lomonosov M et al. Genes Dev 2003; 17:
 results in an increased DNA mutation rate.2. Many DNA repair pathways are lost or deficient in cancer cells. In particular, breast or ovarian tumor cells with hereditary breast cancer associated gene (BRCA) 1 or BRCA2 mutations are HR deficient and so they lack the ability to efficiently repair DSBs.3. Cancer cells continue to grow despite ongoing genomic instability and so the repair pathways they still retain become integral to their survival. Interest in targeting these remaining DNA repair pathways in cancer cells as a selective anti-cancer therapy is developing. References. 1. Khanna K. J Natl Cancer Inst 2000; 92: Bielas JH, Loeb LA. Environ Mol Mutagen 2005; 45: Lomonosov M et al. Genes Dev 2003; 17:")
4
Tipi di lesione al DNA e meccanismi di riparazione
Tipo di lesione: Single- strand breaks (SSBs) Double- strand breaks (DSBs) Addotti ingombranti O6- alchilguanina Inserzioni & delezionsi Mismatch repair Base excision repair Recombinational repair Riparazione diretta Meccanismo di riparazione: Nucleotide- excision repair DNA damage can occur in several different forms, including SSBs or double-strand breaks (DSBs).1 In higher eukaryotes, genomic stability is essential for healthy functioning and survival. DNA damage may induce mutations and can lead to cell death via apoptosis.2 Therefore, several repair mechanisms have evolved to maintain the integrity of the genome.1 Base excision repair (BER) is a key pathway in the repair of SSBs and is reliant on the enzyme poly(ADP-ribose) polymerase (PARP).1 For DSB repair, there are two predominant pathways: Homologous recombination (HR) that involves a protein kinase, ataxia-telangiectasia mutated (ATM) Non-homologous end-joining (NHEJ) that requires DNA-dependent protein kinase (DNA-PK).2 HR is the most accurate mechanism for repairing DSBs, whereas NHEJ is rarely error-free.2 Abbreviations on slide: AGT, O(6)-alkylguanine-DNA alkyltransferase; ATM, ataxia telangiectasia mutated; MLH1, MutL homolog; MSH2, MutS homolog; XP, xeroderma pigmentosum References 1. Jackson SP, Bishop CL. Drug Discovery World 2003; Fall: 2. Jackson SP. Biochem Soc Trans 2001; 29: HR NHEJ Enzimi di riparazione: PARP ATM DNA-PK XP, polimerasi MSH2, MLH1 AGT
 Double- strand. breaks (DSBs) Addotti. ingombranti. O6- alchilguanina. Inserzioni. & delezionsi. Mismatch. repair. Base. excision. repair. Recombinational. repair. Riparazione diretta. Meccanismo. di riparazione: Nucleotide- excision. repair. DNA damage can occur in several different forms, including SSBs or double-strand breaks (DSBs).1. In higher eukaryotes, genomic stability is essential for healthy functioning and survival. DNA damage may induce mutations and can lead to cell death via apoptosis.2 Therefore, several repair mechanisms have evolved to maintain the integrity of the genome.1. Base excision repair (BER) is a key pathway in the repair of SSBs and is reliant on the enzyme poly(ADP-ribose) polymerase (PARP).1. For DSB repair, there are two predominant pathways: Homologous recombination (HR) that involves a protein kinase, ataxia-telangiectasia mutated (ATM) Non-homologous end-joining (NHEJ) that requires DNA-dependent protein kinase (DNA-PK).2. HR is the most accurate mechanism for repairing DSBs, whereas NHEJ is rarely error-free.2. Abbreviations on slide: AGT, O(6)-alkylguanine-DNA alkyltransferase; ATM, ataxia telangiectasia mutated; MLH1, MutL homolog; MSH2, MutS homolog; XP, xeroderma pigmentosum. References. 1. Jackson SP, Bishop CL. Drug Discovery World 2003; Fall: Jackson SP. Biochem Soc Trans 2001; 29: HR. NHEJ. Enzimi. di riparazione: PARP. ATM. DNA-PK. XP, polimerasi. MSH2, MLH1. AGT.")
5
Tse, A. N. et al. Clin Cancer Res 2007;13:1955-1960

6
Fig. 1. DSBs in DNA can arise spontaneously, frequently during replication, or can be induced by exogenous or endogenous agents. The response to DSBs is coordinated by two kinases ATM and ATR, which when activated phosphorylate and activate a plethora of proteins, some of which are shown here. These substrates transduce the DSB response signal to simultaneously stall the cell cycle (predominantly by the activities of CHK1 and CHK2) while triggering either the NHEJ or HR pathways of DNA repair (details of these processes is described in the main text). If the level of DNA damage is assessed to be too great for the cell to effectively process, cell death is induced. This coordinated response ensures that a minimal mutational load is passed to daughter cells once the cell cycle is restarted and mitosis ensues Lord, C. J. et al. Clin Cancer Res 2006;12:

7
Inibitori di ATM, ATR e DNA-PKCS
Non selettivi Caffeina (ATM/ATR) Wortmannina LY294002 ATM-selettivi KU-55933 DNA-PKCS NU-7441
 Wortmannina. LY ATM-selettivi. KU DNA-PKCS. NU")
8
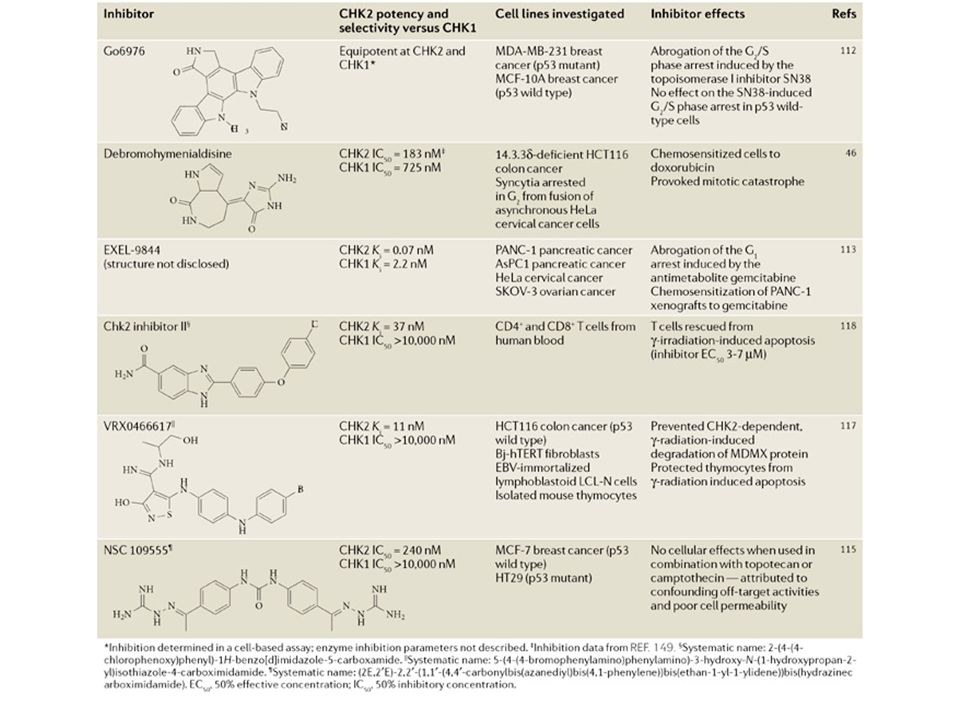
Despite similar nomenclature and some overlapping functions, the roles of CHK1 and CHK2 are distinct, as shown in the table below. In order to further advance therapeutic opportunities and applications, it is essential to clearly define the distinct functions of those two kinases. This will be facilitated by the generation of novel small-molecule inhibitors that are selective for one kinase over the other one.

9
Functions and regulations of CHK1 in the mammalian DNA-damage-response network. CHK1 is activated by a broad spectrum of DNA-damaging agents, including DNA-strand-breaking agents such as ionizing radiation and topoisomerase inhibitors, and those agents that cause replication stress such as ultraviolet light, hydroxyurea and 5-fluorouracil. CHK1 activation that is induced by replication stress is ATR dependent, and its activation by strand-breaking agents is believed to be mainly ATM dependent. Optimal activation of CHK1 also requires other checkpoint proteins such as BRCA1, claspin, the RAD9–RAD1–HUS1 complex and RAD17 (not shown). The role of CHK1 in the S-phase and G2–M checkpoint is best understood and is mediated by phosphorylation of CDC25A and CDC25C, respectively. For simplicity, only one phosphorylation site is indicated for each substrate of CHK1 (though often more than one site is targeted). Other potential CHK1 functions include chromatin remodelling — through the tousled-like kinases (TLK1/2) — replication and DNA repair. UCN-01 is a potent CHK1 inhibitor and has been used as a tool compound to study CHK1-dependent pathways. Dashed lines represent less strong interactions. Red lines represent interactions that are supported by only limited data. Please close this window to return to the main article.

10
MECCANISMI D’AZIONE DI UCN-01
Inibizione isoforme Ca2+-dipendenti della PKC (IC50 = 30 nM) Inibizione isoforme Ca2+-indipendenti della PKC (IC50 ~ 600 nM) Arresto del ciclo cellulare inibizione CDKs aumento della sintesi di p27kip1 e p21waf1 Abolizione del checkpoint in G2
 Inibizione isoforme Ca2+-indipendenti della PKC. (IC50 ~ 600 nM) Arresto del ciclo cellulare. inibizione CDKs. aumento della sintesi di p27kip1 e p21waf1. Abolizione del checkpoint in G2.")
11
Figure 1. Progression through the stages of the cell cycle is regulated by cyclin-dependent kinases (cdks). Cdks are positively regulated by cyclins, levels of which fluctuate throughout the cell cycle, and negatively regulated by the endogenous cdk inhibitors of the INK4a and Cip/Kip families. Arrows indicate sites of action of flavopiridol and UCN-01. (A) Depiction of cell cycle control and phases of the cell cycle where flavopiridol or UCN-01 act. (B) Mechanism by which flavopiridol blocks cell cycle progression. (C) Abrogation of G2 checkpoint by UCN-01. Following DNA damage, the G2 checkpoint is activated, which allows the cell to remain in G2 until all DNA damage is repaired and, thus, to enter M phase with "intact" DNA. However, UCN-01 treatment of DNA-damaged cells abrogates the G2 checkpoint (IC50 ~50 nM), which allows the cells to progress into M prior to completion of DNA repair, leading to apoptosis. The UCN-01 G2 checkpoint abrogation was found to involve Cdc25C, the cdk1 (Cdc2)-activating phosphatase, which is modulated by chk1, a protein kinase directly affected by UCN-01.
. Cdks are positively regulated by cyclins, levels of which fluctuate throughout the cell cycle, and negatively regulated by the endogenous cdk inhibitors of the INK4a and Cip/Kip families. Arrows indicate sites of action of flavopiridol and UCN-01. (A) Depiction of cell cycle control and phases of the cell cycle where flavopiridol or UCN-01 act. (B) Mechanism by which flavopiridol blocks cell cycle progression. (C) Abrogation of G2 checkpoint by UCN-01. Following DNA damage, the G2 checkpoint is activated, which allows the cell to remain in G2 until all DNA damage is repaired and, thus, to enter M phase with intact DNA. However, UCN-01 treatment of DNA-damaged cells abrogates the G2 checkpoint (IC50 ~50 nM), which allows the cells to progress into M prior to completion of DNA repair, leading to apoptosis. The UCN-01 G2 checkpoint abrogation was found to involve Cdc25C, the cdk1 (Cdc2)-activating phosphatase, which is modulated by chk1, a protein kinase directly affected by UCN-01..")
12
Figure 2 | Functions and regulations of CHK2 in the mammalian DNA-damage-response network. CHK2 is mainly activated by DNA-strand-breaking agents such as ionizing radiation and topoisomerase inhibitors through the ATM-dependent pathway. Other checkpoint proteins, such as 53BP1, MDC1 and the MRE11–RAD50–NBS1 complex, might modulate CHK2 activation (not shown). The role of CHK2 in checkpoints is not totally clear (see main text for details), although it has been shown to phosphorylate CDC25A in vitro, which inhibits its activity. The role of CHK2 in DNA-damage-induced apoptosis is better established. It operates through both p53-dependent and p53-independent — through PML and E2F — pathways. Pifithrin- can block p53-dependent transcription and apoptosis, and has been used as a tool to validate the p53-dependent pathway as a radio/chemosensitization target. CHK2 has also been shown to phosphorylate the BRCA1 protein and might modulate the role of BRCA1 in DNA repair. Red lines represent interactions that are supported by only limited data.
. The role of CHK2 in checkpoints is not totally clear (see main text for details), although it has been shown to phosphorylate CDC25A in vitro, which inhibits its activity. The role of CHK2 in DNA-damage-induced apoptosis is better established. It operates through both p53-dependent and p53-independent — through PML and E2F — pathways. Pifithrin- can block p53-dependent transcription and apoptosis, and has been used as a tool to validate the p53-dependent pathway as a radio/chemosensitization target. CHK2 has also been shown to phosphorylate the BRCA1 protein and might modulate the role of BRCA1 in DNA repair. Red lines represent interactions that are supported by only limited data..")
14
PARP-1 è un enzima chiave nella riparazione di single-strand DNA breaks
DNA damage PARP PAR chains are degraded via PARG Binds directly to SSBs Repaired DNA PARP-1 is found in the cell’s nucleus and is a key component of BER, the DNA SSB repair process.1 PARP-1 activation is an immediate cellular response to metabolic, chemical, or radiation-induced DNA SSB damage.2 PARP-1 mediates the repair of SSBs via the activation and recruitment of repair enzymes. Firstly, PARP-1 detects and signals the presence of an SSB by binding to the DNA adjacent to the damage. Once bound, PARP-1 catalyzes the cleavage of the coenzyme nicotinamide adenine dinucleotide (NAD+) into nicotinamide and ADP-ribose to produce highly charged branched chains of poly(ADP-ribose) (PAR). These activities of PARP-1 serve to recruit other repair enzymes, such as DNA ligase III (LigIII) and DNA polymerase beta (polβ), and scaffolding proteins such as x-ray repair complementing gene 1 (XRCC1), to the site of damage, where together the enzymes repair the damaged DNA.2 DNA polymerase uses the complementary strand of DNA as a template to fix the SSB, and DNA ligase creates the final phosphodiester bond to fully repair the DNA. After repair, the PAR chains are degraded via PAR glycohydrolase (PARG). References 1. Hoeijmakers JH. Nature 2001; 411: 2. D’Amours D et al. J Biochem 1999; 342: NAD+ Nicotinamide +pADPr Repair enzymes PAR Once bound to damaged DNA, PARP modifies itself producing large branched chains of PAR
 into nicotinamide and ADP-ribose to produce highly charged branched chains of poly(ADP-ribose) (PAR). These activities of PARP-1 serve to recruit other repair enzymes, such as DNA ligase III (LigIII) and DNA polymerase beta (polβ), and scaffolding proteins such as x-ray repair complementing gene 1 (XRCC1), to the site of damage, where together the enzymes repair the damaged DNA.2 DNA polymerase uses the complementary strand of DNA as a template to fix the SSB, and DNA ligase creates the final phosphodiester bond to fully repair the DNA. After repair, the PAR chains are degraded via PAR glycohydrolase (PARG). References. 1. Hoeijmakers JH. Nature 2001; 411: D’Amours D et al. J Biochem 1999; 342: NAD+ Nicotinamide +pADPr. Repair enzymes. PAR. Once bound to damaged DNA, PARP modifies itself producing large branched chains of PAR.")
15
L’inibizione di PARP-1 aumenta le lesioni a doppio filamento nel DNA
DNA SSB XRCC1 LigIII PNK 1 pol β PARP Inhibition of PARP-1 prevents recruitment of repair factors to repair SSB Inhibition of PARP-1 activity prevents the recruitment of DNA repair enzymes and leads to failure of SSB repair and accumulation of SSBs. During the S-phase of the cell cycle, the replication fork is arrested at the site of an SSB, which then degenerates into a DSB. In normal cells, this triggers activation of the HR pathway to repair the DSB.1 Abbreviation on slide: PNK 1, polynucleotide kinase 1 Reference 1. Helleday T et al. Cell Cycle 2005; 4: Replication (S-phase) DNA DSB
 DNA DSB.")
16
Fig. 1. Mechanism of PARP action
Fig. 1. Mechanism of PARP action. DNA-damaging agents, such as chemotherapeutics, radiation, or replication errors, activate PARP, resulting in poly(ADP-ribose)–branched chains attached to DNA, recruiting associated repair proteins and cell cycle checkpoint mediators. This cascade may lead to cell cycle arrest while the cell commits to either DNA repair or apoptosis. Overactivation of PARP will lead to NAD+ depletion and necrotic cell death. PARP inhibition is thought to impair DNA repair function, leading to cellular dysfunction and death, and may also affect other PARP mediated DNA modulating effects. Ratnam, K. et al. Clin Cancer Res 2007;13:
–branched chains attached to DNA, recruiting associated repair proteins and cell cycle checkpoint mediators. This cascade may lead to cell cycle arrest while the cell commits to either DNA repair or apoptosis. Overactivation of PARP will lead to NAD+ depletion and necrotic cell death. PARP inhibition is thought to impair DNA repair function, leading to cellular dysfunction and death, and may also affect other PARP mediated DNA modulating effects. Ratnam, K. et al. Clin Cancer Res 2007;13:")
17
Fig. 3. Schematic drawing to explain PARP activation induced by N3-MeA
Fig. 3. Schematic drawing to explain PARP activation induced by N3-MeA. In the presence of low levels of MPG activity a high number of N3-MeA persists in DNA. During DNA replication, DNA polymerase is blocked by the methyl adduct. N3-MeA adducts located in the unreplicated single strand DNA are highly unstable and give rise to apurinic sites which are not further processed and repaired by BER [8]. The resulting DNA nicks, in proximity of stalled replication fork, eventually generate double strand breaks that may cause PARP activation

18
Fig. 4. PARP inhibition and sensitization to DNA damage induced by topoisomerase I poisons. The figure illustrates a simplified model on how PARP inhibition may increase cytotoxcity of topoisomerase I inhibitors. PARP-1 and PARP-2 act as carriers of (ADP-ribose) polymers, which bind to specific domains of topoisomerase I, favouring the resealing of strand breaks by the ligase activity of topoisomerase I. In the presence of topoisomerase I inhibitor double strand breaks are generated when the replication fork encounters a cleavable complex. During this process, accelerated DNA re-ligation mediated by (ADP-ribose) polymers would counteract the action of the antitumor drug. Addition of PARP inhibitor may impair strand break re-joining mediated by topoisomerase I itself, increasing DNA double strand break formation.
 polymers, which bind to specific domains of topoisomerase I, favouring the resealing of strand breaks by the ligase activity of topoisomerase I. In the presence of topoisomerase I inhibitor double strand breaks are generated when the replication fork encounters a cleavable complex. During this process, accelerated DNA re-ligation mediated by (ADP-ribose) polymers would counteract the action of the antitumor drug. Addition of PARP inhibitor may impair strand break re-joining mediated by topoisomerase I itself, increasing DNA double strand break formation..")
19
Effetto Selettivo dell’inibizione di PARP-1 in cellule tumorali con mutazioni di BRCA1 or BRCA2
DSB in DNA Normal cell BRCA-deficient cancer cell DSB repaired effectively via HR pathway Deficient HR pathway – DSB not repaired When PARP-1 is inhibited, SSBs collapse replication forks leading to an increase in DSBs.1,2 In normal replicating cells, DSBs are repaired by the HR pathway. BRCA1 or BRCA2 are a vital part of the HR mechanism as without these proteins, the replication fork cannot be restarted.3 In cells that are deficient in HR repair (eg breast or ovarian cancer cells with a BRCA1 or BRCA2 deficiency), the inhibition of PARP activity leads to an increase in DSBs that cannot be repaired by this mechanism. DSBs can induce genomic instability and cell-death mechanisms such as apoptosis.2 In addition, many of these DSBs would be repaired by other, error-prone mechanisms, leading to large numbers of aberrations and this in turn would also ultimately lead to cell death. The effect of PARP inhibition would be highly specific to tumor cells that have a HR deficiency (eg BRCA1 or BRCA2 deficiency). This results in selective cell killing and an increased therapeutic ratio; normal cells have a working HR repair pathway and so would not be affected in this way. References 1. Bryant HE et al. Nature 2005; 434: 2. Helleday T et al. Cell Cycle 2005; 4: 3. Lomonosov M et al. Genes Dev 2003; 17: Cell survival Cancer cell death
, the inhibition of PARP activity leads to an increase in DSBs that cannot be repaired by this mechanism. DSBs can induce genomic instability and cell-death mechanisms such as apoptosis.2 In addition, many of these DSBs would be repaired by other, error-prone mechanisms, leading to large numbers of aberrations and this in turn would also ultimately lead to cell death. The effect of PARP inhibition would be highly specific to tumor cells that have a HR deficiency (eg BRCA1 or BRCA2 deficiency). This results in selective cell killing and an increased therapeutic ratio; normal cells have a working HR repair pathway and so would not be affected in this way. References. 1. Bryant HE et al. Nature 2005; 434: Helleday T et al. Cell Cycle 2005; 4: Lomonosov M et al. Genes Dev 2003; 17: Cell survival. Cancer cell death.")
20
a | Cell division cycle 25 (CDC25) phosphatases dephosphorylate and activate cyclin-dependent kinase (CDK)–cyclin complexes, thus allowing catalysis and substrate phosphorylation. WEE1 and MYT1 kinases phosphorylate CDK on tyrosine 15 and threonine 14 of CDK1. Phosphorylation by the CDK-activating kinase (CAK) is required for further activation of CDK–cyclin complexes. For simplicity, an orange P represents T14 and Y15 phosphorylation by WEE1 and MYT1, a blue P represents T161 phosphorylation by CAK. b | Although initial studies suggested a specific role for each CDC25 phosphatase at defined stages of the cell cycle, the current model is that CDC25A, B and C are all involved in phosphorylating CDK–cyclin complexes, such as CDK2–cyclin E at the G1–S transition or CDK1–cyclin B at the entry into mitosis. c | CDC25A, B and C control entry and progression into mitosis. CDC25B is thought to be responsible for the initial activation of CDK1–cyclin B at the centrosome that contributes to microtubule network reorganization and mitotic spindle assembly. Nuclear translocation leads to an auto-amplification process (bold arrows) of CDC25s that then fire the bulk of CDK1–cyclin B complexes and trigger mitosis.
 is required for further activation of CDK–cyclin complexes. For simplicity, an orange P represents T14 and Y15 phosphorylation by WEE1 and MYT1, a blue P represents T161 phosphorylation by CAK. b | Although initial studies suggested a specific role for each CDC25 phosphatase at defined stages of the cell cycle, the current model is that CDC25A, B and C are all involved in phosphorylating CDK–cyclin complexes, such as CDK2–cyclin E at the G1–S transition or CDK1–cyclin B at the entry into mitosis. c | CDC25A, B and C control entry and progression into mitosis. CDC25B is thought to be responsible for the initial activation of CDK1–cyclin B at the centrosome that contributes to microtubule network reorganization and mitotic spindle assembly. Nuclear translocation leads to an auto-amplification process (bold arrows) of CDC25s that then fire the bulk of CDK1–cyclin B complexes and trigger mitosis..")
21
A schematic view of our current knowledge of the phosphorylation sites for cell division cycle 25 (CDC25) phosphatases. Most of these phosphorylations are located within the N-terminal regulatory domain of the proteins (C-terminal catalytic domains are shown as cream boxes; not to scale). CDC25A, CDC25B and CDC25C are phosphorylated by multiple kinases that regulate their activity, interactions with proteins, and intracellular localizations by the modulation of nuclear export sequence (NES) and nuclear localization signal (NLS). For simplicity, many additional putative cyclin-dependent kinase (CDK)–cyclin phosphorylation sites are not depicted here. Degradation by the SCF TRCP or by the APC/C–ubiquitin (Ub)-dependent degradation pathways is also controlled by the indicated phosphorylation events.
. CDC25A, CDC25B and CDC25C are phosphorylated by multiple kinases that regulate their activity, interactions with proteins, and intracellular localizations by the modulation of nuclear export sequence (NES) and nuclear localization signal (NLS). For simplicity, many additional putative cyclin-dependent kinase (CDK)–cyclin phosphorylation sites are not depicted here. Degradation by the SCF TRCP or by the APC/C–ubiquitin (Ub)-dependent degradation pathways is also controlled by the indicated phosphorylation events..")
22
a, b | Upon DNA damage, cell division cycle 25 (CDC25) proteins are inhibited through various mechanisms, including checkpoint kinase-dependent degradation or cytoplasmic sequestration through binding (see Ref. 23 for a review). The inhibitory kinases WEE1 and MYT1 are activated by checkpoint kinases. CDK–cyclin complexes are in turn maintained in their inactive state and the cell remains arrested in G2 phase. c | CDC25B level is central to the control of entry into mitosis after DNA damage. Together with PLK1 activity (which is required for WEE1 inhibition), CDC25B accumulation through a mechanism that is still unclear78 is required to allow entry into mitosis when damage has been repaired162. Increased levels of CDC25B protein found in cancer might therefore facilitate checkpoint exit and increase genomic instability77
. The inhibitory kinases WEE1 and MYT1 are activated by checkpoint kinases. CDK–cyclin complexes are in turn maintained in their inactive state and the cell remains arrested in G2 phase. c | CDC25B level is central to the control of entry into mitosis after DNA damage. Together with PLK1 activity (which is required for WEE1 inhibition), CDC25B accumulation through a mechanism that is still unclear78 is required to allow entry into mitosis when damage has been repaired162. Increased levels of CDC25B protein found in cancer might therefore facilitate checkpoint exit and increase genomic instability77.")
23
Cell division cycle 25 (CDC25) proteins reported to be overexpressed in primary tumour samples from patients with breast96, prostate84, ovarian163, endometrial104, colorectal88, 89, oesophageal91, 92, 94, 112, thyroid96, 97, 98, laryngeal99, gastric101 and hepatocellular cancers100, glioma107, neuroblastoma106 or non-Hodgkin lymphoma109. Percentages of tumours in which CDC25A, CDC25B or CDC25C proteins are overexpressed are indicated, or in the case of overexpression reported in more than one study, a range of percentages is given. The cut-off mark for inclusion was overexpression in >10% patients. Whether (Y) or not (N) CDC25 overexpression was linked to clinicopathological features, including tumour grade or stage, metastases, depth of invasion, residual or recurrent disease, or disease-free survival is marked in yellow boxes beside percentages. Cases for which several studies reported contradictory prognostic values are marked by a (C), and studies in which clinicopathological features were not assessed are indicated by an (?).
 or not (N) CDC25 overexpression was linked to clinicopathological features, including tumour grade or stage, metastases, depth of invasion, residual or recurrent disease, or disease-free survival is marked in yellow boxes beside percentages. Cases for which several studies reported contradictory prognostic values are marked by a (C), and studies in which clinicopathological features were not assessed are indicated by an ( )..")
Presentazioni simili

>")











>")





