Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Classificazione dei livelli strutturali in proteine

2
Classificazione dei livelli strutturali in proteine
Struttura primaria (sequenza amminoacidica)
")
3
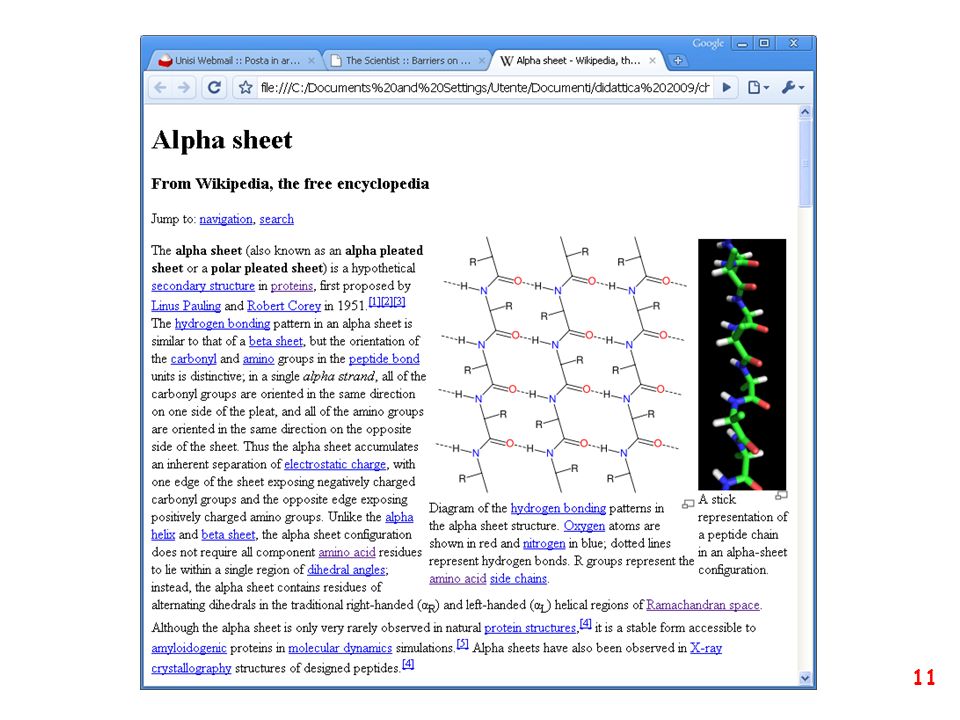
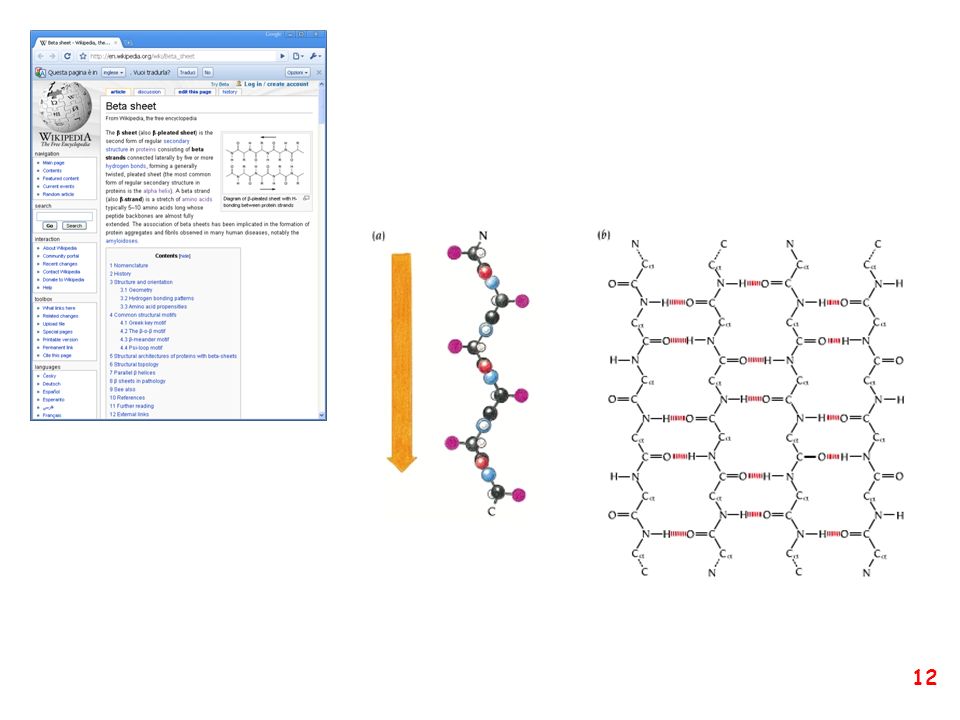
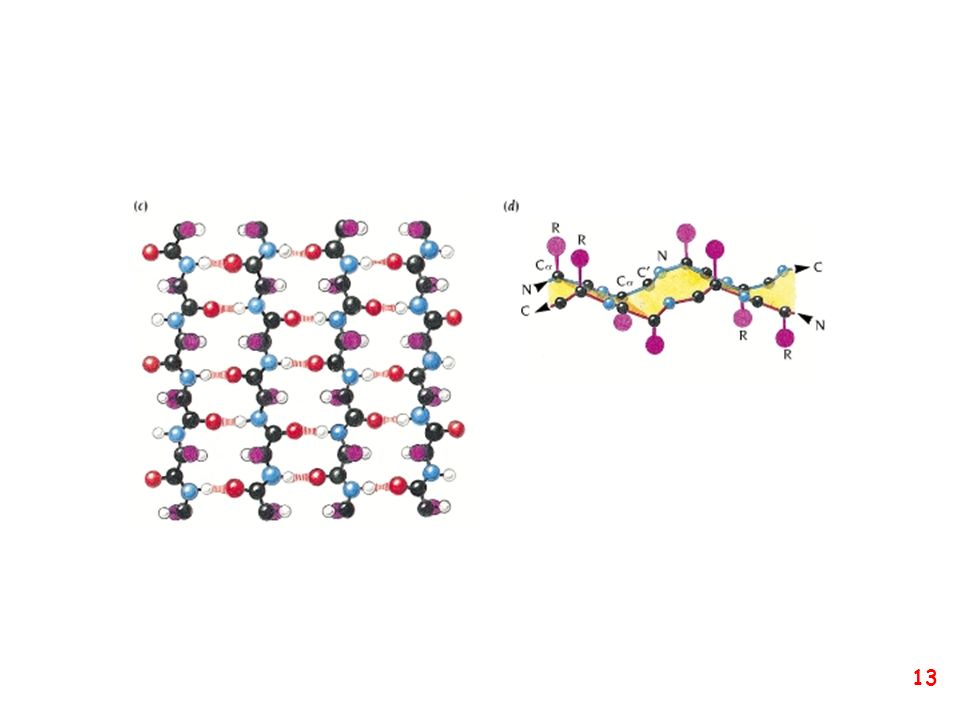
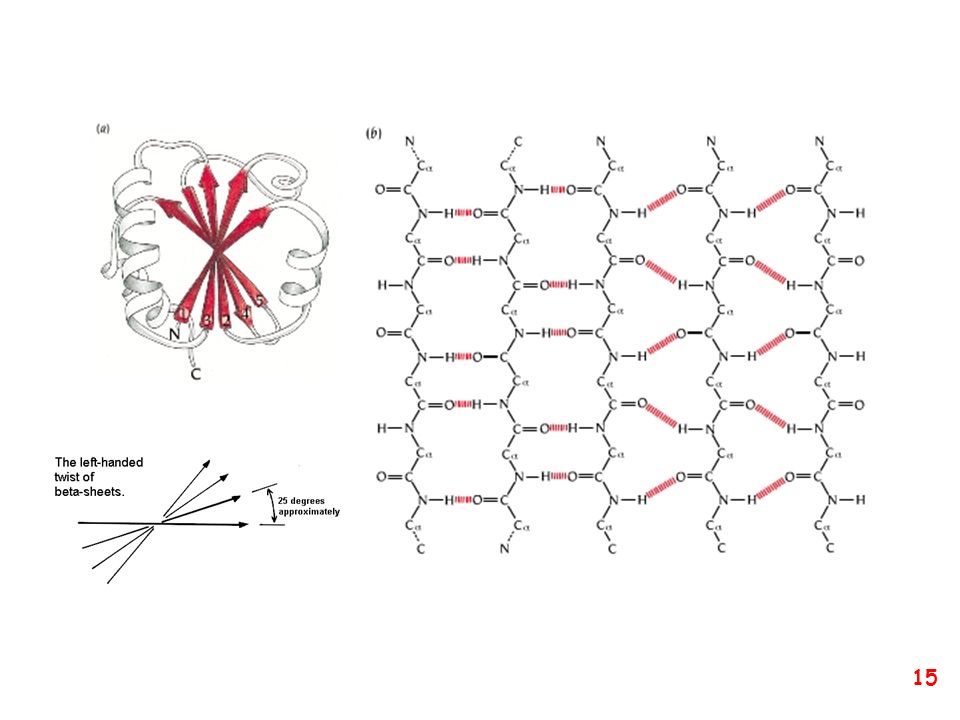
struttura secondaria: la forma della catena principale della proteina
(protein backbone)
")
4
DIAGRAMMA DI RAMACHANDRAN Gopalasamudram Narayana Iyer Ramachandran
8 October April 2001 Journal of Molecular Biology - 1963

5
DIAGRAMMA DI RAMACHANDRAN
φ ψ 180 -180 φ ψ

6
Conformazione delle catene laterali
Torsion Axes and Dihedral Angles of the side chain of Lysine The sample amino acid Lysine has four torsion axes within its side chain. The torsion axes are symbolized as arrows, the dihedral angles are labeled chi1 to chi4.

7
eliche α in α-helix hydrogen bonds between carbonyl i and amide i+4
An α-helix in ultra-high-resolution electron density contours, with O atoms in red, N atoms in blue, and hydrogen bonds as green dotted lines (PDB file 2NRL, 17-32).
.")
8
Folding mechanism of alpha helices

9
φ ψ 180 -180 eliche α, ma non solo

19
anse

20
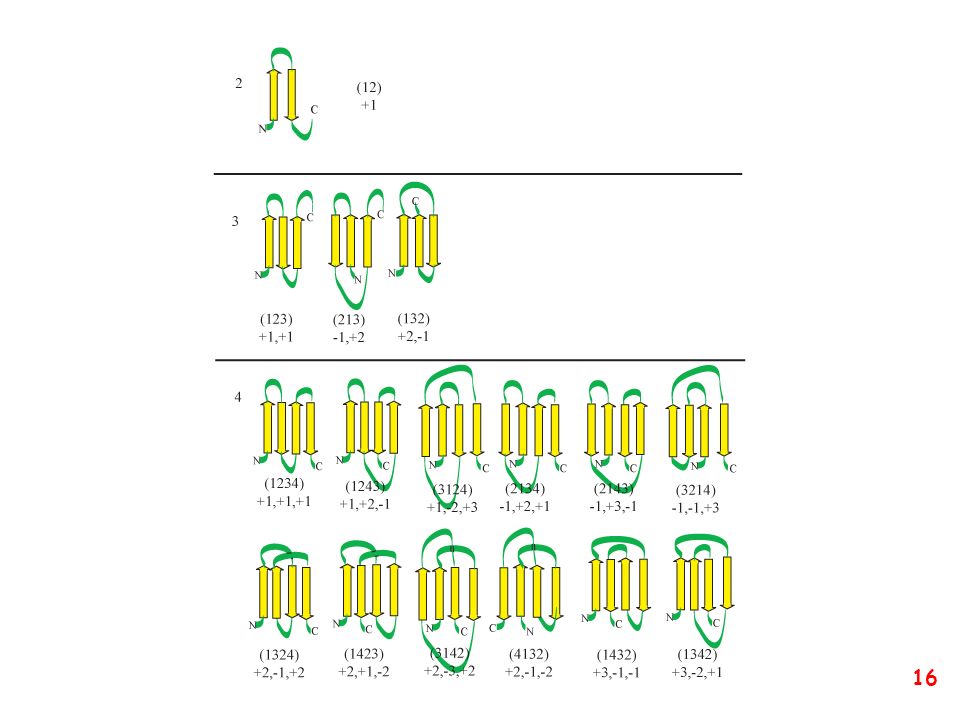
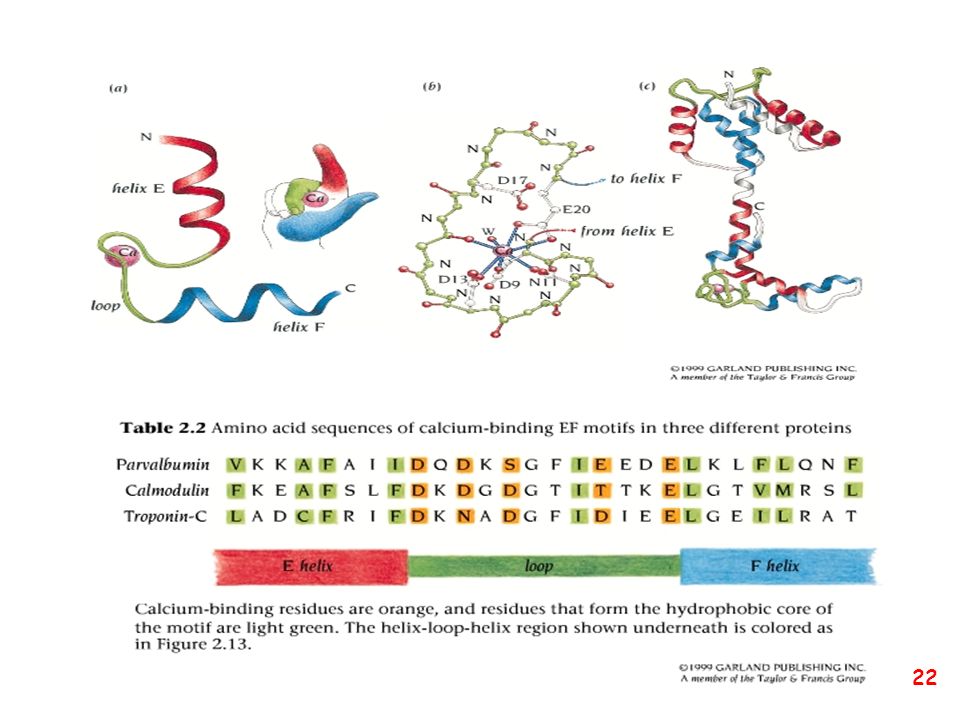
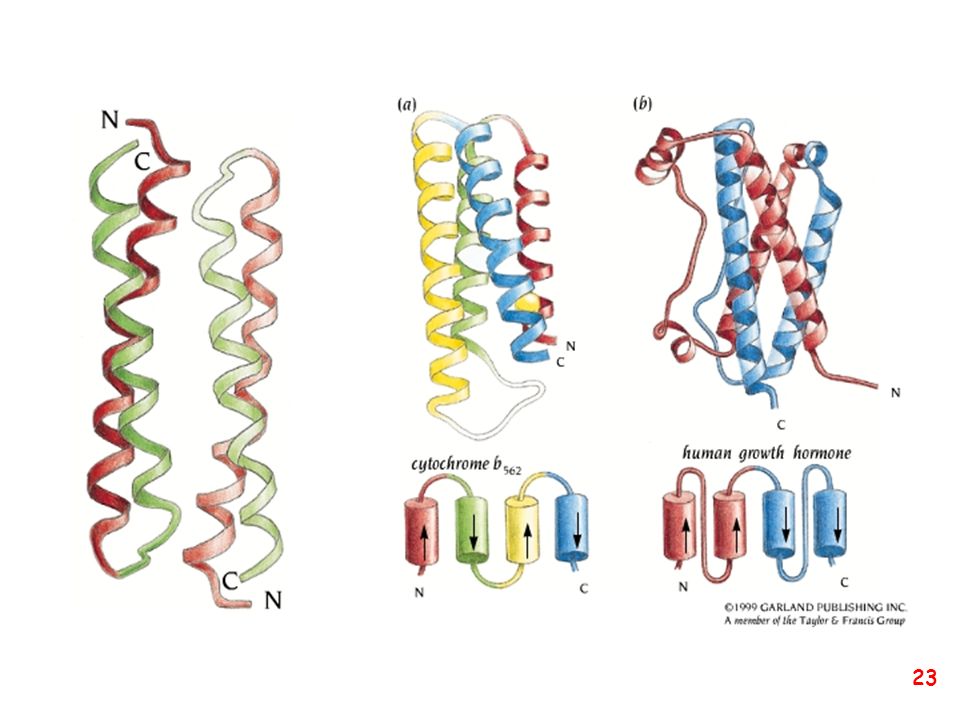
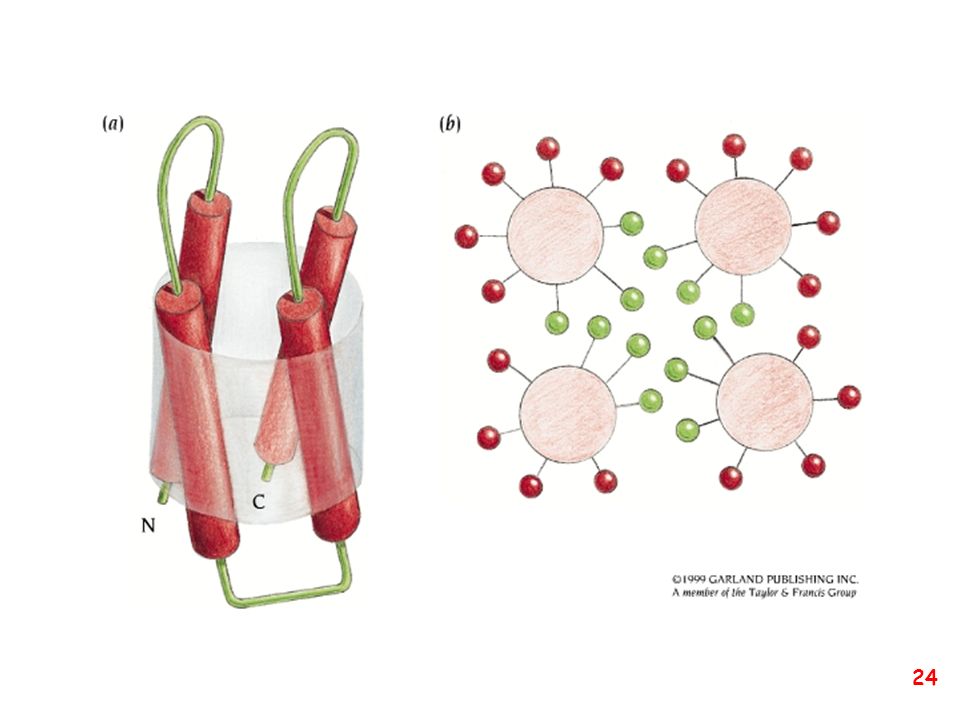
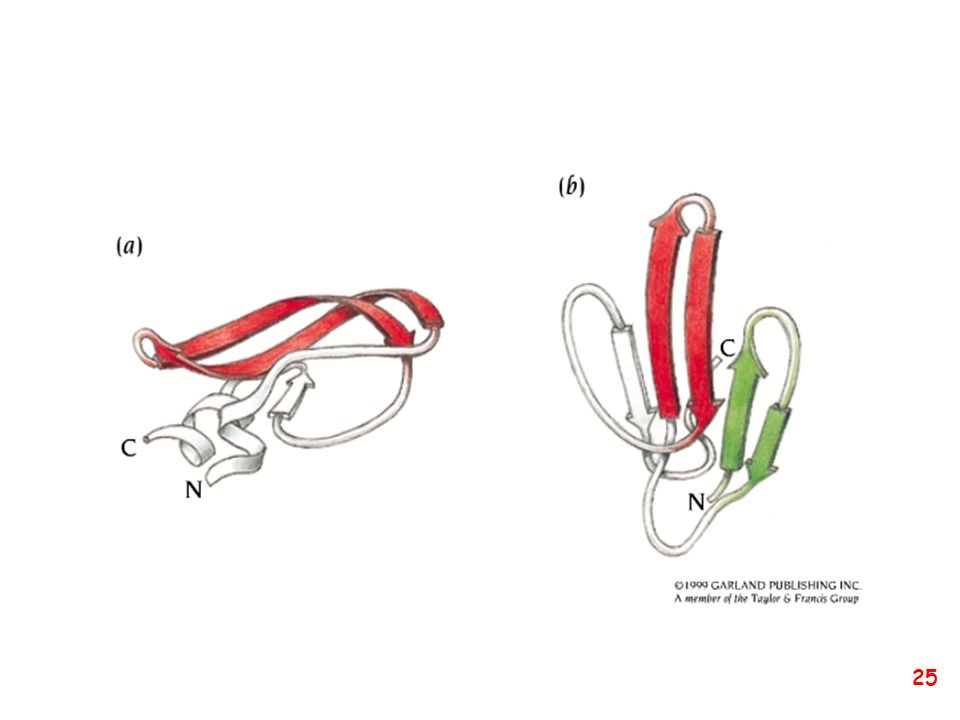
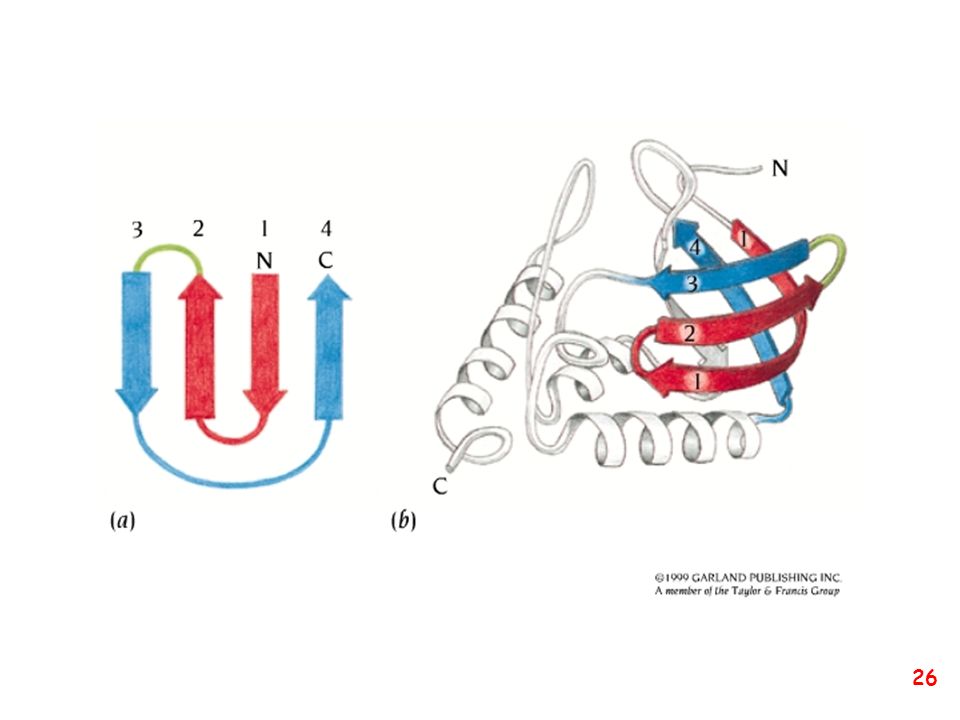
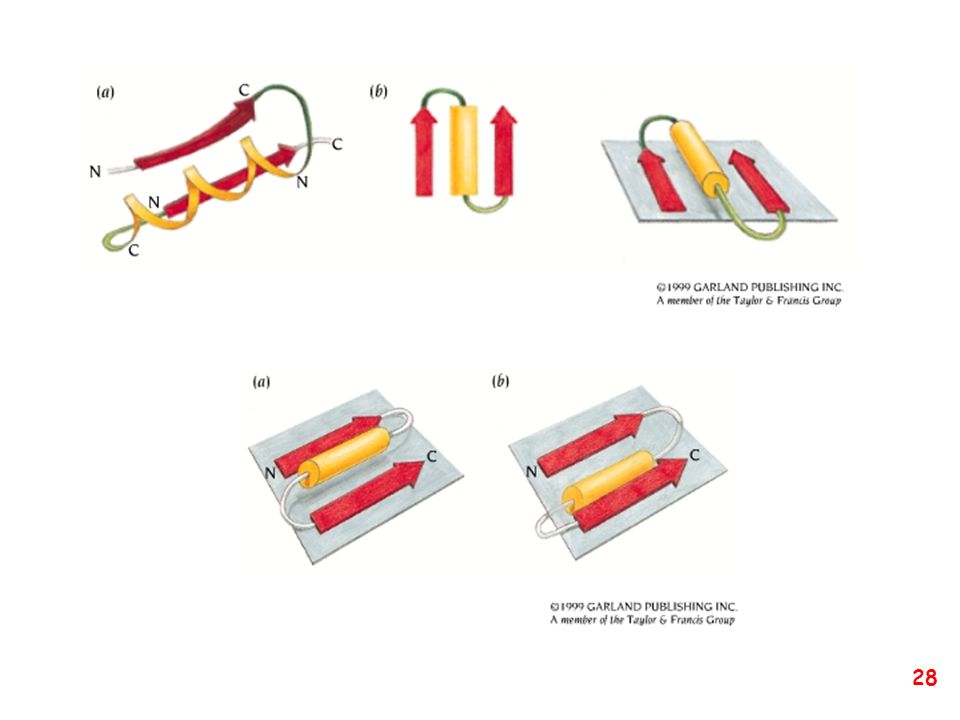
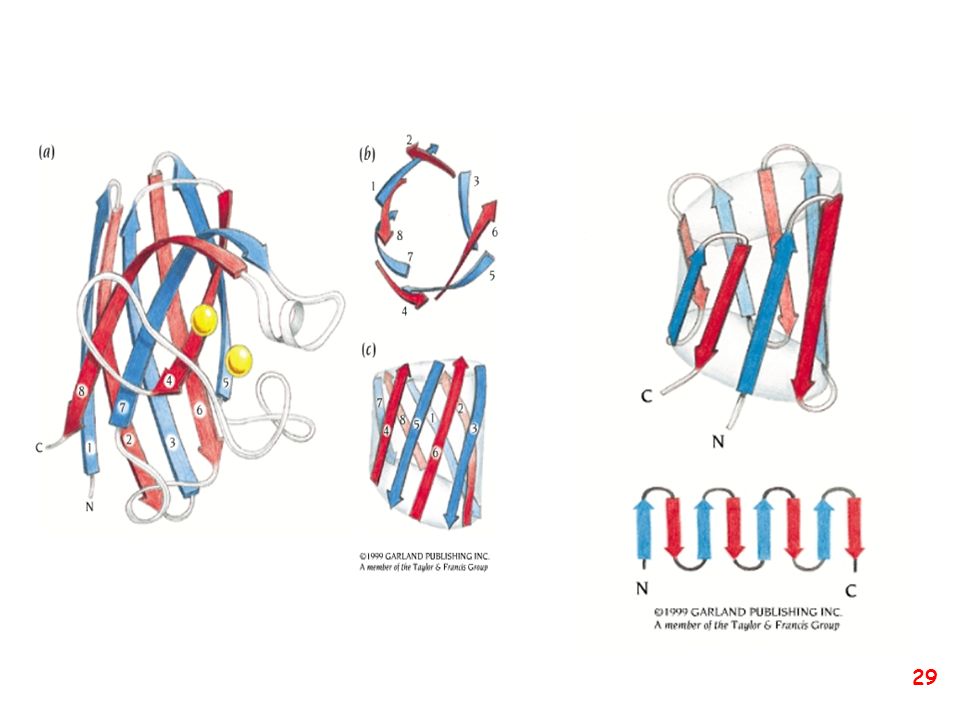
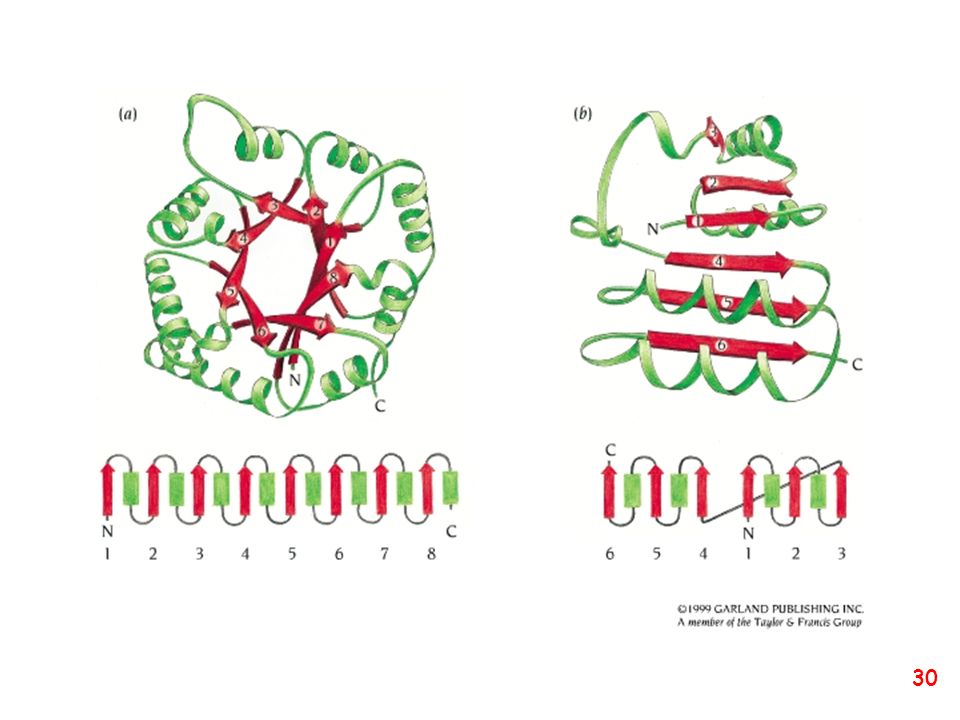
Strutture supersecondarie o motivi
(elementi di struttura secondaria uguali o diversi si combinano) a: bab b: ripiegamento a forcina a: motivo aa d: barile b e: barile a/b (e)
 a: bab. b: ripiegamento a forcina. a: motivo aa. d: barile b. e: barile a/b. (e)")
31
(subunità avente stabilità strutturale ed una propria funzione)
Domini strutturali (subunità avente stabilità strutturale ed una propria funzione) dominio che lega il gliceraldeide-3-fosfato dominio che lega il NAD+ gliceraldeide-3-fosfato deidrogenasi
 dominio che lega il. gliceraldeide-3-fosfato. dominio che lega il NAD+ gliceraldeide-3-fosfato deidrogenasi.")
33
Enzima bifunzionale Piruvato chinasi PRA-isomerasi IGP-sintetasi

34
Struttura quaternaria
Eteromultimeri Omomultimeri

35
Intrinsically Unstructured Proteins (IUPs)
Set of keywords used: Intrinsically disordered proteins

36
Implications… Cell size constraints? The natively unstructured state is a simple and elegant solution adopted by evolution to avoid large protein, genome and cell sizes TiBS, 2003, 28, 81 Schematic representation of dimers (a) unstable (disordered) and (b) stable (ordered) monomers. Although in both cases the interface area between the monomers is the same, the size of the ordered monomer is much larger compared with the disordered example
 unstable (disordered) and (b) stable (ordered) monomers. Although in both cases the interface area between the monomers is the same, the size of the ordered monomer is much larger compared with the disordered example.")
37
Some Structural Features
Combination of low overall hydrophobicity and relative high net charge under physiological conditions A combination of low mean hydrophobicity and high net charge preclude the formation of a hydrophobic cluster and promote an extended conformation Blue squares=275folded Red circle=91 IUPs Green circles=130 Predicted IUPs Cyan circle=242 Homologues of IUPs Proteins, 2000, 41,415

38
Table 2 Aminoacid Frequencies of Ordered and Disordered Proteins
Some Structural Features… IUPs have a distinctive aa composition Table 2 Aminoacid Frequencies of Ordered and Disordered Proteins IUPs are enriched in S,P,E,K (disorder promoting) and depleted in W, Y,F,C,I, L, N (order promoting) TiBS, 2002, 527
 and depleted in W, Y,F,C,I, L, N (order promoting) TiBS, 2002, 527.")
39
Taxonomy is the practice and science of classification
Linnaeus[5] (1735) 2 kingdoms Haeckel[6] (1866) 3 kingdoms Chatton[7] (1925) 2 groups Copeland[8] (1938) 4 kingdoms Whittaker[2] (1969) 5 kingdoms Woese [9][10] (1977,1990) 3 domains Animalia Eukaryote Eukarya Vegetabilia Plantae Protoctista Fungi (not treated) Protista Procaryote Monera Archaea Bacteria
 2 kingdoms. Haeckel[6] (1866) 3 kingdoms. Chatton[7] (1925) 2 groups. Copeland[8] (1938) 4 kingdoms. Whittaker[2] (1969) 5 kingdoms. Woese [9][10] (1977,1990) 3 domains. Animalia. Eukaryote. Eukarya. Vegetabilia. Plantae. Protoctista. Fungi. (not treated) Protista. Procaryote. Monera. Archaea. Bacteria.")
40
Phylogenetic Tree of All Life

43
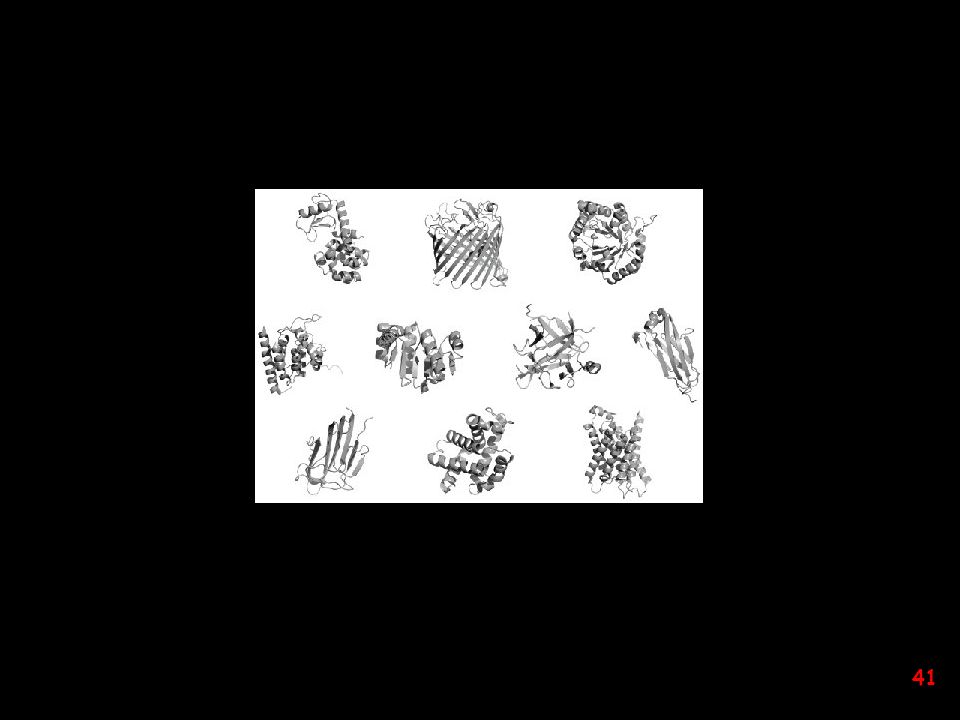
estimated number of protein folds: ~ 2000 (?)
This three-dimensional map of the protein universe shows the distribution in space of the 500 most common protein folds as represented by spheres. The spheres, which are colored according to classification, reveal four distinct classes. From It is conceivable that, of the primordial peptides, those containing fragments with high helix and/or strand propensity found their way to fold into small alpha, beta, and alpha plus beta structures," Kim says. "The alpha slash beta fold structures do not appear until proteins of sufficient size rose through evolution and the formation of supersecondary structural units became possible. estimated number of protein folds: ~ 2000 (?)
")
44
Statistics of new folds in PDB

45
Top Folds in Yeast Transcriptome

46
TIM barrel nelle strutture depositate nel PDB

47
ENERGIA TOTALE CONFORMAZIONALE
E=Ea+Er+Ees+El+Et+Ef+EH+EHf Ea attrazione Er repulsione Ees potenziale elettrostatico El variazione di lunghezze di legame Et variazione di angoli di legame Ef potenziale torsionale EH legame ad idrogeno Ehf interazione idrofobica

48
Protein structure dynamics

49
ENERGIA TOTALE CONFORMAZIONALE
E=Ea+Er+Ees+El+Et+Ef+EH+EHf

51
Molecular Dynamics simulation

52
Molecular Dynamics simulation

54
Nel paradosso di Levinthal una proteina di 100 residui raggiungerebbe la struttura nativa attraverso una ricerca casuale nello spazio conformazionale in 1087 s. Ogni aa con tre possibilità di y e f: 3200 conformazioni diverse. Tempo minimo di interconversione s 3200/ circa 1087 s

55
Illustration of the main driving force behind protein structure formation. In the compact fold (to the right), the hydrophobic amino acids (shown as black spheres) are in general shielded from the solvent. Ipotetico meccanismo di ripiegamento di una proteina con due domini: formazione di elementi di struttura secondaria locali con formazione dei domini e loro assemblaggio finale. La struttura terziaria è raggiunta in pochi secondi.

57
This is the E. coli major cold shock protein, CspA
This is the E. coli major cold shock protein, CspA. This folding pathway is completely fictitious. The structure of CspA was solved by Hermann Schindelin and coworkers (PNAS, 91: , 1994). The coordinates for the cold shock protein (ID code "1mjc") may be retrieved from the Brookhaven Protein Data Bank. Below is a step by step presentation.
. The coordinates for the cold shock protein (ID code 1mjc ) may be retrieved from the Brookhaven Protein Data Bank. Below is a step by step presentation.")
60
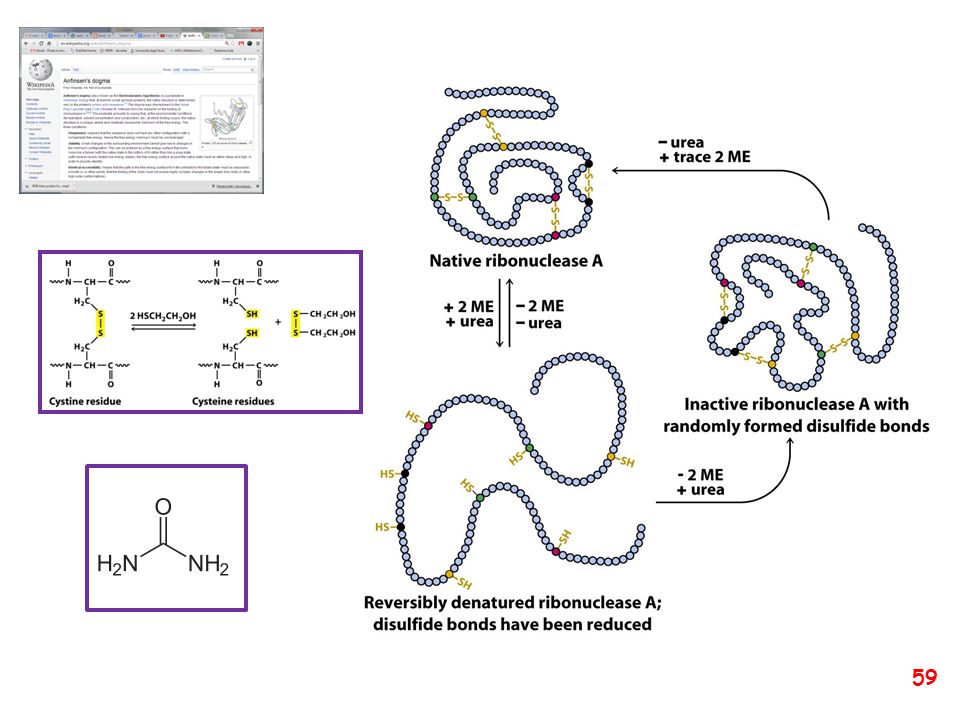
thiol-disulfide oxidoreductase

61
Chaperone-assisted protein folding
The unfolded polypeptide enters the central cavity of chaperonin, where it folds. The hydrolysis of several ATP molecules is required for chaperonin function. The three dimensional proteins structure is shown in Figure. The lines on the chaperonin cylinder are to represent the 7 identical GroEL subunits that make up each ring. Not shown is the end cap composed of GroES subunits.

62
E. coli chaperonin (GroE)
The core structure of chaperonin consists of two identical rings composed of seven GroEL subunits. Unfolded prteins bind to the central cavity. Bound ATP molecules can be identified by their red oxygen atoms (spacefill). The quaternary structure is shown from (a) the side, and (b) the top. [PDB 1DER] (c) During folding, the size of the central cavity of one of the rings increases and the end is capped by a protein containing seven GroES subunits. [PDB 1AON]. Highlighted in green is one of the GroEL subunits.
. The quaternary structure is shown from (a) the side, and (b) the top. [PDB 1DER] (c) During folding, the size of the central cavity of one of the rings increases and the end is capped by a protein containing seven GroES subunits. [PDB 1AON]. Highlighted in green is one of the GroEL subunits.")
63
The enclosed cavity is the site of protein folding
Misfolded proteins in the cavity are given about 20 seconds to refold. After 20 seconds, they are expelled either refolded successfully, or given another chance to enter the same or a different chaperon complex to try again A misfolded protein will be kept in “solitary confinement” until it has reformed correctly.

64
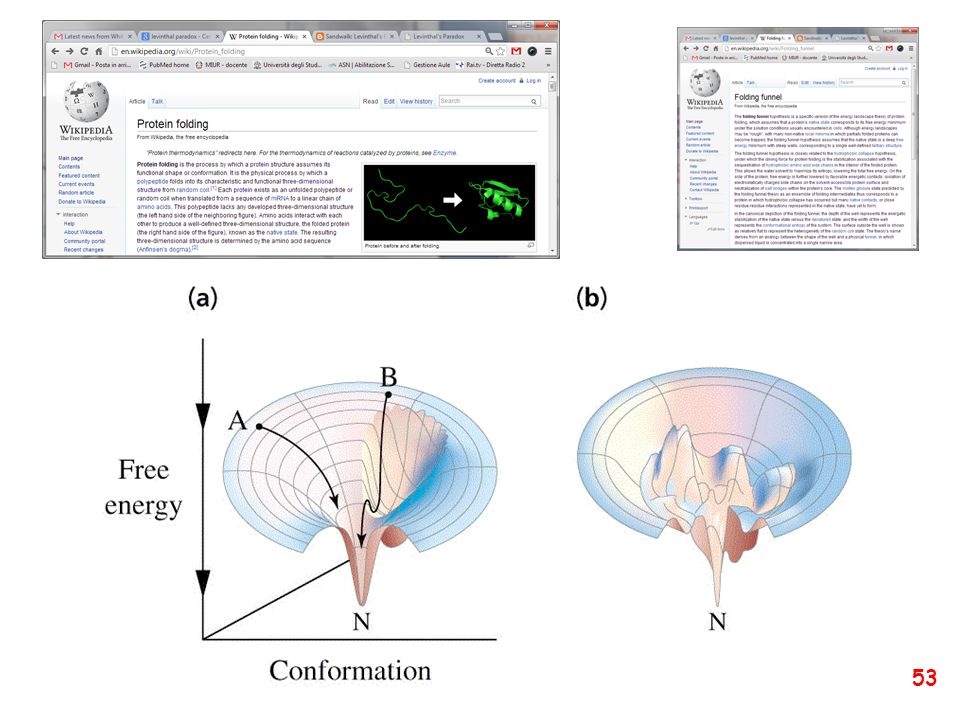
Schematic of the folding energy landscape of a protein molecule where the energy of the protein is displayed as a function of the topological arrangements of the atoms. The multiple states of the unfolded protein located at the top fall into a folding funnel consisting of an almost infinite number of local minima, each of which describes possible folding arrangements in the protein. Most of these states represent transient folding intermediates in the process of attaining the correct native fold. Some of these intermediates retain a more stable structure such as the molten globule, whereas other local minima act as folding traps irreversibly capturing the protein in a misfolded state. Christian P. Schultz Illuminating folding intermediates Nature Structural Biology 7, (2000)
")
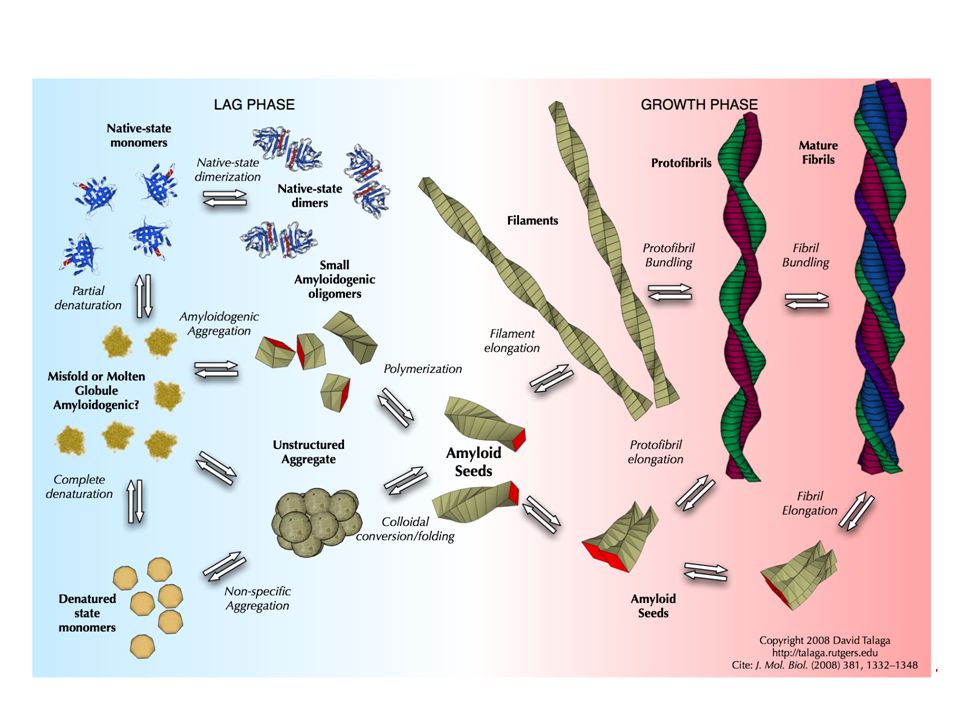
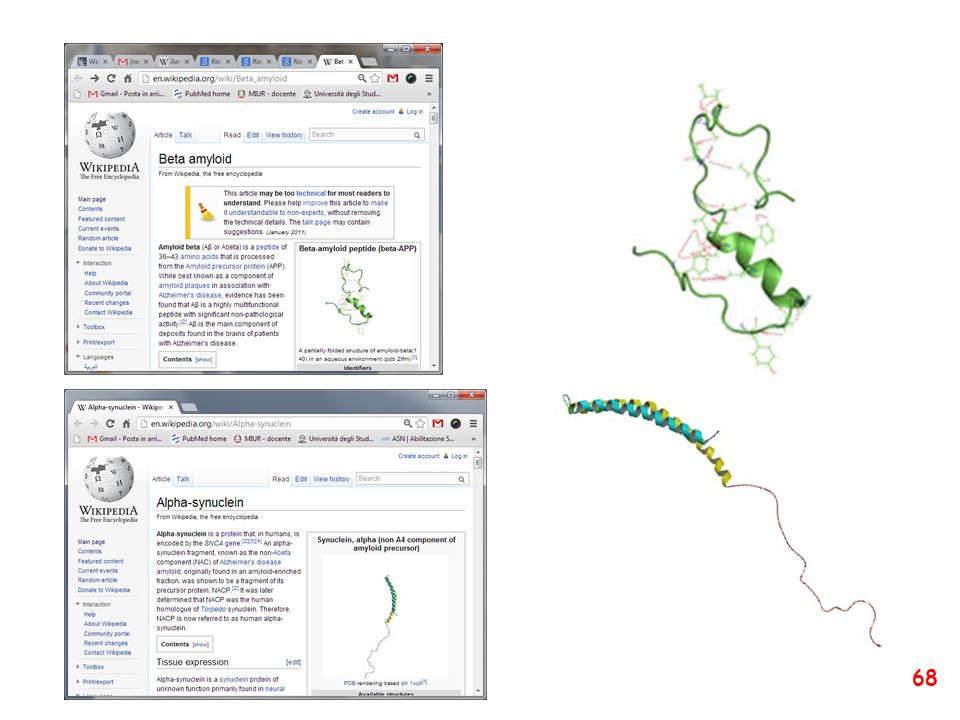
66
amiloidosi The ability of proteins to change conformation is the essence of the amyloidoses — in these diseases, the proteins have converted into the ‘primordial’ structure rather than remaining in their evolved states.

70
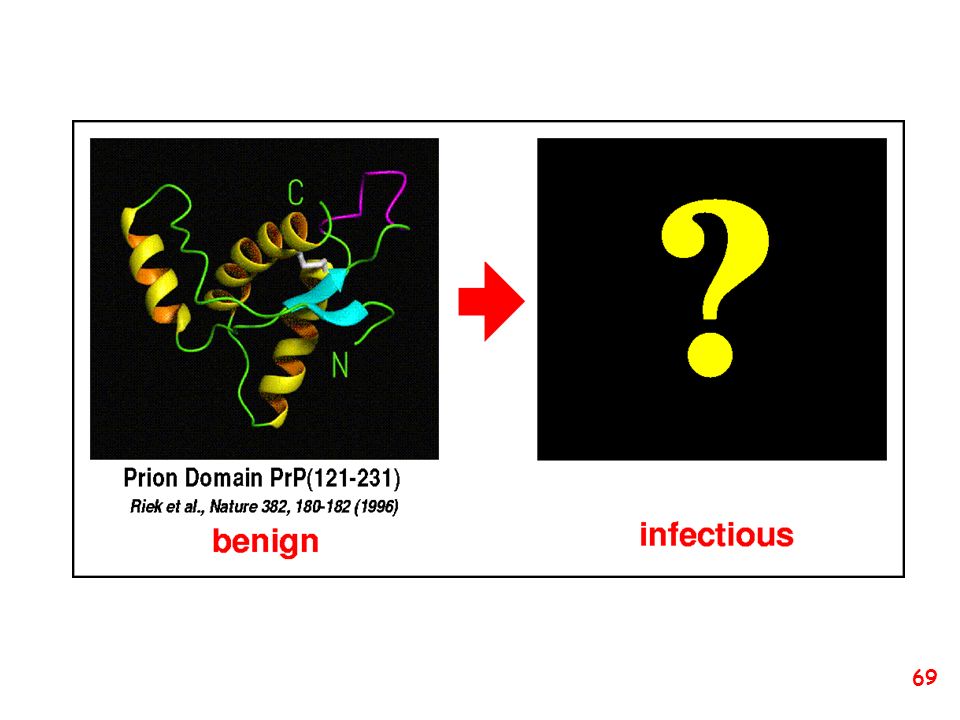
Proposed three-dimensional structure (a) PrPC and (b) PrPSc
The structure of the normal prion protein, PrPC, is characterised by four -helices. Conversion of PrPC to the disease-associated form of PrPSc results in the loss of two of the helical structures (shown shaded in brown), which are converted to linear structures known as -sheets. It is this conversion that is associated with the aquisition of prion infectivity.
, which are converted to linear structures known as -sheets. It is this conversion that is associated with the aquisition of prion infectivity.")
74
Anemia falciforme mutazione E/V in posizione 6 nella emoglobina

Presentazioni simili