Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
I dispositivi medici e di protezione individuale specifici e la segnalazione degli incidenti
Istituti Fisioterapici Ospitalieri Roma 15, 16 e 17 maggio 2006 Nicoletta Jannitti, Farmacista Dirigente I.F.O.

2
Sommario I dispositivi medici (dm): cenni di normativa
Alcuni dm specifici per allestimento e somministrazione farmaci citotossici I dispositivi di protezione individuale (DPI): cenni di normativa I DPI specifici per allestimento e somministrazione farmaci citotossici La segnalazione di incidenti con dispositivi medici
: cenni di normativa. I DPI specifici per allestimento e somministrazione farmaci citotossici. La segnalazione di incidenti con dispositivi medici.")
3
I dispositivi medici sono regolamentate da tre direttive principali:
direttiva 90/385/CEE sui dispositivi medici impiantabili attivi direttiva 93/42/CEE sui dispositivi medici direttiva 98/79/CE sui dispositivi medici diagnostici in vitro (IVD)
")
4
Il mercato unico entro il 31 dicembre 1992
Necessaria una nuova tecnica di regolamentazione che: definisse solo i requisiti essenziali generali; riducesse il controllo delle autorità pubbliche prima dell’immissione nel mercato di un prodotto; integrasse tecniche moderne di valutazione della conformità (approccio globale). Necessario uno snellimento del processo decisionale Per realizzare l’obiettivo della creazione di un mercato unico entro il 31 dicembre 1992, si rendeva necessaria una nuova tecnica di regolamentazione che: definisse solo i requisiti essenziali generali; riducesse il controllo delle autorità pubbliche prima dell’immissione nel mercato di un prodotto; integrasse tecniche moderne di valutazione della conformità (approccio globale); Parallelamente il processo decisionale doveva essere adeguato: le direttive di armonizzazione tecnica potevano essere adottate a maggioranza qualificata in seno al Consiglio e non più all’unanimità.
. Necessario uno snellimento del processo decisionale. Per realizzare l’obiettivo della creazione di un mercato unico entro il 31 dicembre 1992, si rendeva necessaria una nuova tecnica di regolamentazione che: definisse solo i requisiti essenziali generali; riducesse il controllo delle autorità pubbliche prima dell’immissione nel mercato di un prodotto; integrasse tecniche moderne di valutazione della conformità (approccio globale); Parallelamente il processo decisionale doveva essere adeguato: le direttive di armonizzazione tecnica potevano essere adottate a maggioranza qualificata in seno al Consiglio e non più all’unanimità.")
5
Il nuovo approccio Nuova tecnica regolamentare: a) l’armonizzazione legislativa si limita ai requisiti essenziali; b) le specifiche tecniche dei prodotti che rispondono ai requisiti essenziali vengono definite in norme armonizzate; c) l’applicazione di norme armonizzate o di altro genere rimane volontaria; d) i prodotti fabbricati nel rispetto delle norme armonizzate sono ritenuti conformi ai corrispondenti requisiti essenziali. Affinchè il sistema possa funzionare devono essere soddisfatte due condizioni: in primo luogo le norme armonizzate devono fornire un livello di protezione garantito rispetto ai requisiti essenziali fissati nelle direttive (garantire la qualità del prodotto conforme); in secondo luogo le autorità competenti devono vigilare sulla sicurezza (o su altri requisiti) sul loro territorio. Questa è una condizione necessaria per assicurare la fiducia reciproca tra stati membri. Si prevede inoltre una procedura per l'applicazione della clausola di salvaguardia, che consenta di contestare la conformità di un prodotto o eventuali carenze delle norme armonizzate . Risoluzione del Consiglio, del 7 maggio 1985, relativa ad una nuova strategia in materia di armonizzazione tecnica e normalizzazione Istituita una nuova tecnica regolamentare che ha fissato principi di seguito enunciati: l’armonizzazione legislativa si limita ai requisiti essenziali che i prodotti immessi nel mercato della Comunità devono rispettare per poter circolare liberamente all’interno della Comunità stessa; le specifiche tecniche dei prodotti che rispondono ai requisiti essenziali fissati nelle direttive vengono definite in norme armonizzate, la cui elaborazione è affidata agli organi competenti in materia di normalizzazione industriale l’applicazione di norme armonizzate o di altro genere rimane volontaria e il fabbricante può sempre applicare altre specifiche tecniche per soddisfare i requisiti previsti i prodotti fabbricati nel rispetto delle norme armonizzate sono ritenuti conformi ai corrispondenti requisiti essenziali. (le amministrazioni sono tenute a riconoscere ai prodotti fabbricati secondo le norme armonizzate una presunzione di conformità ai requisiti fondamentali stabiliti dalla direttiva)
 le specifiche tecniche dei prodotti che rispondono ai requisiti essenziali vengono definite in norme armonizzate; c) l’applicazione di norme armonizzate o di altro genere rimane volontaria; d) i prodotti fabbricati nel rispetto delle norme armonizzate sono ritenuti conformi ai corrispondenti requisiti essenziali. Affinchè il sistema possa funzionare devono essere soddisfatte due condizioni: in primo luogo le norme armonizzate devono fornire un livello di protezione garantito rispetto ai requisiti essenziali fissati nelle direttive (garantire la qualità del prodotto conforme); in secondo luogo le autorità competenti devono vigilare sulla sicurezza (o su altri requisiti) sul loro territorio. Questa è una condizione necessaria per assicurare la fiducia reciproca tra stati membri. Si prevede inoltre una procedura per l applicazione della clausola di salvaguardia, che consenta di contestare la conformità di un prodotto o eventuali carenze delle norme armonizzate . Risoluzione del Consiglio, del 7 maggio 1985, relativa ad una nuova strategia in materia di armonizzazione tecnica e normalizzazione. Istituita una nuova tecnica regolamentare che ha fissato principi di seguito enunciati: l’armonizzazione legislativa si limita ai requisiti essenziali che i prodotti immessi nel mercato della Comunità devono rispettare per poter circolare liberamente all’interno della Comunità stessa; le specifiche tecniche dei prodotti che rispondono ai requisiti essenziali fissati nelle direttive vengono definite in norme armonizzate, la cui elaborazione è affidata agli organi competenti in materia di normalizzazione industriale. l’applicazione di norme armonizzate o di altro genere rimane volontaria e il fabbricante può sempre applicare altre specifiche tecniche per soddisfare i requisiti previsti. i prodotti fabbricati nel rispetto delle norme armonizzate sono ritenuti conformi ai corrispondenti requisiti essenziali. (le amministrazioni sono tenute a riconoscere ai prodotti fabbricati secondo le norme armonizzate una presunzione di conformità ai requisiti fondamentali stabiliti dalla direttiva)")
6
Direttive di Nuovo Approccio Elementi standard
Campo di applicazione Immissione in mercato e messa in servizio Requisiti essenziali Libera circolazione Presunzione di conformità Clausola di salvaguardia Valutazione di conformità Organismi notificati Marcatura CE Coordinamento dell’attuazione Recepimento e disposizioni transitorie

7
sono direttive di Nuovo Approccio
direttiva 90/385/CEE sui dispositivi medici impiantabili attivi direttiva 93/42/CEE sui dispositivi medici direttiva 98/79/CE sui dispositivi medici diagnostici in vitro (IVD) direttiva 89/686/CEE sui dispositivi di protezione individuale sono direttive di Nuovo Approccio
 direttiva 89/686/CEE sui dispositivi di protezione individuale. sono direttive di Nuovo Approccio.")
8
Direttive di Nuovo Approccio La marcatura CE
La marcatura CE è una dichiarazione della persona responsabile che i prodotti: sono conformi ai requisiti essenziali di tutte le direttive applicabili; sono stati sottoposti ad una procedura di valutazione della conformità prevista dalle direttive stesse. I requisiti essenziali fissati dalle direttive del nuovo approccio possono sovrapporsi o integrarsi a vicenda. Il prodotto può essere immesso nel mercato e messo in servizio solo se è conforme alle disposizioni di tutte le direttive applicabili e quando la valutazione della conformità viene eseguita secondo le modalità di tutte le direttive applicabili.

9
Dispositivi medici D. Lgs
Dispositivi medici D.Lgs. 24 febbraio 1997, n°46, attuazione della Direttiva 93/42/CEE Art.4 (Requisiti essenziali) I dispositivi devono soddisfare i pertinenti requisiti essenziali prescritti nell’allegato I in considerazione della loro destinazione. Art. 6 (Rinvio alle norme) Si presume conforme ai requisiti essenziali di cui all’art. 4 il dispositivo fabbricato in conformità delle norme armonizzate comunitarie e delle norme nazionali che le recepiscono.
 I dispositivi devono soddisfare i pertinenti requisiti essenziali prescritti nell’allegato I in considerazione della loro destinazione. Art. 6 (Rinvio alle norme) Si presume conforme ai requisiti essenziali di cui all’art. 4 il dispositivo fabbricato in conformità delle norme armonizzate comunitarie e delle norme nazionali che le recepiscono.")
10
Marcatura CE per i dm D.Lgs. 24 febbraio 1997, n°46,
Le procedure necessarie all’apposizione delle marcatura CE sono diverse a seconda della classe di rischio di appartenenza. Per le classi superiori alla I non sterile, interviene anche un organismo notificato.

11
Destinazione d’uso D.Lgs. 24 febbraio 1997, n°46,
È definita dal fabbricante. È ciò che determina la classe di rischio. La marcatura CE indica che il dm è idoneo all’impiego secondo la destinazione d’uso dichiarata.

12
Destinazione d’uso e responsabilità dell’utilizzatore D. Lgs
Destinazione d’uso e responsabilità dell’utilizzatore D.Lgs. 24 febbraio 1997, n°46, Il dm è progettato, fabbricato e testato in funzione della sua destinazione d’uso. L’impiego di un dm al di fuori della destinazione d’uso prevista ricade sotto la responsabilità dell’utilizzatore.

13
Direttive di Nuovo Approccio Il fabbricante
E’ la persona responsabile della progettazione e della fabbricazione di un prodotto al fine di immetterlo nel mercato della Comunità per suo conto. E’ il solo ed unico responsabile della conformità del proprio prodotto alle direttive applicabili.

14
Direttive di Nuovo Approccio Il fabbricante
E’ il solo e unico responsabile della conformità del proprio prodotto alle direttive applicabili, sia che abbia progettato e fabbricato il prodotto personalmente, sia che l’abbia solo immesso nel mercato a suo nome. Ha la responsabilità di a) progettare e fabbricare il prodotto nel rispetto dei requisiti essenziali fissati nella o nelle direttive; b) eseguire la valutazione della conformità secondo le procedure fissate nella o nelle direttive. Può utilizzare prodotti finiti, pezzi già pronti, o componenti o può subappaltare le operazioni che gli competono, ma deve comunque mantenere il controllo globale e deve disporre delle competenze necessarie per assumersi la responsabilità del prodotto.
 progettare e fabbricare il prodotto nel rispetto dei requisiti essenziali fissati nella o nelle direttive; b) eseguire la valutazione della conformità secondo le procedure fissate nella o nelle direttive. Può utilizzare prodotti finiti, pezzi già pronti, o componenti o può subappaltare le operazioni che gli competono, ma deve comunque mantenere il controllo globale e deve disporre delle competenze necessarie per assumersi la responsabilità del prodotto.")
15
I dispositivi medici D. Lgs
I dispositivi medici D.Lgs. 24 febbraio 1997, n°46, attuazione della Direttiva 93/42/CEE Definizione: qualsiasi strumento, apparecchio, impianto, sostanza (…) destinato dal fabbricante ad essere impiegato nell’uomo a scopo di diagnosi, prevenzione, controllo, terapia (…) il quale prodotto non eserciti l’azione principale, cui è destinato, con mezzi farmacologici (…)
 destinato dal fabbricante ad essere impiegato nell’uomo a scopo di diagnosi, prevenzione, controllo, terapia (…) il quale prodotto non eserciti l’azione principale, cui è destinato, con mezzi farmacologici (…)")
16
I dispositivi medici Classe I : dispositivi a basso rischio (non invasivi, invasivi a breve termine in orifizi naturali, strumenti chirurgici riutilizzabili,...) Classe II a: dispositivi a rischio medio (non invasivi ad es. per contatto con cute lesa, per somministrazione di liquidi; invasivi a breve t. in orifizi naturali diversi da Classe I; invasivi chir. per uso temporaneo,...) Classe II b: dispositivi a rischio medio-alto (non invasivi ad es. sacche x sangue, materiali x contatto con derma leso; invasivi chir. a breve termine;...) Classe III: dispositivi a rischio alto (dm destinati a contatto con organi vitali; impiantabili; chir. a lungo termine, ...) classificazione in base a destinazione d'uso, durata d'impiego, rischio potenziale derivante dall'utilizzo
 Classe II a: dispositivi a rischio medio (non invasivi ad es. per contatto con cute lesa, per somministrazione di liquidi; invasivi a breve t. in orifizi naturali diversi da Classe I; invasivi chir. per uso temporaneo,...) Classe II b: dispositivi a rischio medio-alto (non invasivi ad es. sacche x sangue, materiali x contatto con derma leso; invasivi chir. a breve termine;...) Classe III: dispositivi a rischio alto (dm destinati a contatto con organi vitali; impiantabili; chir. a lungo termine, ...) classificazione in base a destinazione d uso, durata d impiego, rischio potenziale derivante dall utilizzo.")
17
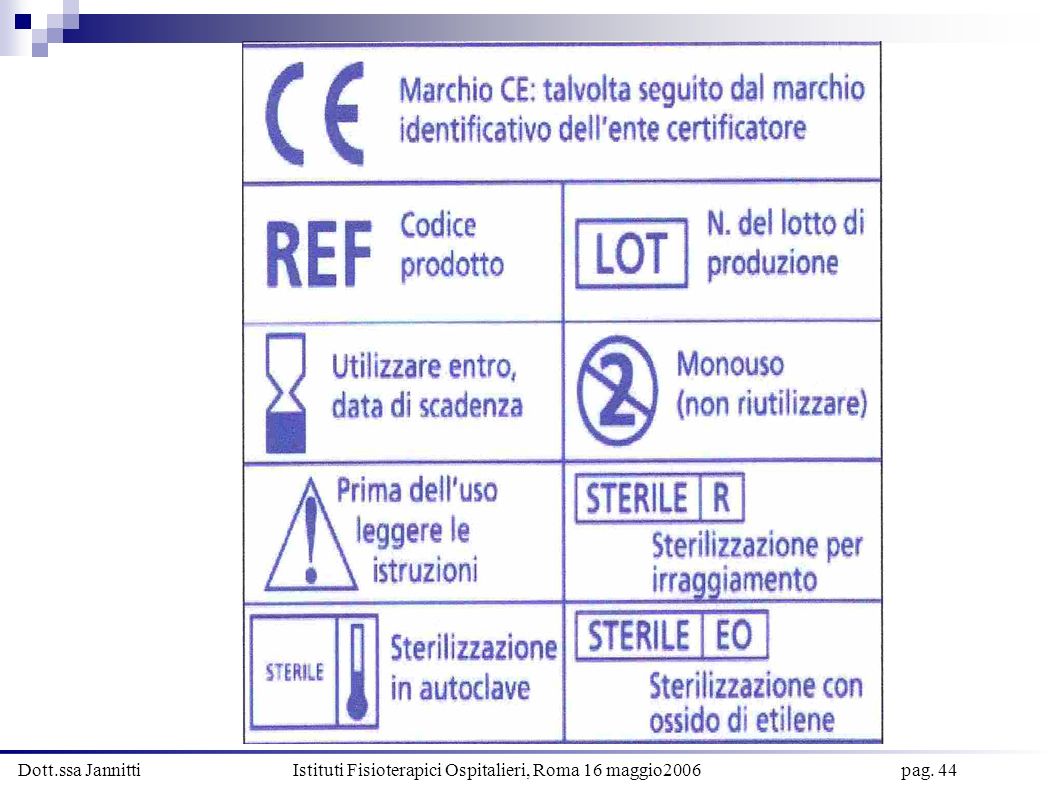
(…) 13. Informazioni fornite dal fabbricante
REQUISITI ESSENZIALI Allegato 1 Lgs. 24 febbraio 1997, n°46 (…) 13. Informazioni fornite dal fabbricante Ogni dm deve essere corredato dalle necessarie informazioni per garantirne un’utilizzazione sicura e per consentire di identificare il fabbricante (…) Le informazioni sono costituite dalle indicazioni riportate su etichetta e istruzioni per l’uso. Tutti i dispositivi devono contenere nell’imballaggio le istruzioni per l’uso. In via eccezionale non necessarie per dm classe I e IIa, qualora sia possibile garantire un’utilizzazione sicura senza dette istruzioni. Se del caso, le informazioni vanno fornite sotto forma di simboli (…)
 13. Informazioni fornite dal fabbricante. Ogni dm deve essere corredato dalle necessarie informazioni per garantirne un’utilizzazione sicura e per consentire di identificare il fabbricante (…) Le informazioni sono costituite dalle indicazioni riportate su etichetta e istruzioni per l’uso. Tutti i dispositivi devono contenere nell’imballaggio le istruzioni per l’uso. In via eccezionale non necessarie per dm classe I e IIa, qualora sia possibile garantire un’utilizzazione sicura senza dette istruzioni. Se del caso, le informazioni vanno fornite sotto forma di simboli (…)")
18
I dispositivi medici per allestimento e somministrazione dei farmaci citotossici

19
Spike equalizzatori di pressione per ricostituzione
Spike con attacco luer lock per somministrazione Pompe elastomeriche

21
Spike per la ricostituzione di farmaci citostatici in polvere e il prelievo da flaconi multidose Mini-Spike V Chemo BBraun Perforatore a doppia via Linea “liquidi” con valvola automatica di chiusura e filtro particelle (5 m) Linea ingresso aria per compensazione P con filtro 0.2 m a ritenzione aerosol Aggancio siringa idoneo a cono luer e luer lock Tappo chiusura a scatto Senza PVC LATEX FREE (scheda tecnica)
 Linea ingresso aria per compensazione P con filtro 0.2 m a ritenzione aerosol. Aggancio siringa idoneo a cono luer e luer lock. Tappo chiusura a scatto. Senza PVC. LATEX FREE (scheda tecnica)")
23
Spike per la ricostituzione di farmaci citostatici in polvere e il prelievo da flaconi multidose Mini-Spike V Chemo rosso BBraun Istruzioni per l’uso (…) Conservare tra i 15°C e i 25°C, umidità relativa tra 50% e 60%.
 Conservare tra i 15°C e i 25°C, umidità relativa tra 50% e 60%.")
24
Puntale perforatore con presa d’aria (filtro 3 m) richiudibile
Valvola autorichiudente attivata da luer maschio (valvola “Clearlink) Senza PVC LATEX FREE
 Senza PVC. LATEX FREE.")
25
Spike per (la preparazione) e la somministrazione di soluzioni infusionali EMC3481 Baxter
Puntale perforatore con presa d’aria (filtro 3 m) richiudibile Valvola autorichiudente attivata da luer maschio (valvola “Clearlink) Senza PVC LATEX FREE
 richiudibile. Valvola autorichiudente attivata da luer maschio (valvola Clearlink) Senza PVC. LATEX FREE.")
26
How it works Baxter’s new needle free system
• When a luer is attached to the Clearlink, the luer compresses the pierced silicone septum opening the aperture and forcing the centre post down thus allows transfer of fluids. FLUIDS Inlet housing Gland Aperture opening Free transfer of fluids Centre post Outlet housing Central post forced down FLUIDS • When the luer is removed, the septum rises to seal off the fluid channel and maintain the integrity of the IV line.

27
Spike per (la preparazione) e la somministrazione di soluzioni infusionali EMC3481 Baxter Istruzioni per l’uso Precauzioni (…) Non accedere alla Clearlink con un ago. Raccomandato l’uso di collegamenti luer lock. Se si usa un collegamento luer semplice, inserirlo nella valvola usando un movimento deciso di spinta e torsione. Non lasciare il luer semplice incustodito.
 Non accedere alla Clearlink con un ago. Raccomandato l’uso di collegamenti luer lock. Se si usa un collegamento luer semplice, inserirlo nella valvola usando un movimento deciso di spinta e torsione. Non lasciare il luer semplice incustodito.")
28
Easypump La pompa elastomerica
è un sistema di infusione esterno, non programmabile, che permette l’infusione continua di un farmaco, non dipendente dalla gravità.

29
Easypump Descrizione
Doppia membrana elastomerica Guscio esterno protettivo in morbido PVC Valvola di riempimento Linea di infusione in PVC con: - morsetto - filtro particellare - filtro eliminazione aria - valvola regolazione flusso

30
Descrizione materiali
Membrana interna in miscela di polimeri Membrana intermedia in lattice (No contatto farmaco o paziente) Guscio esterno protettivo in morbido PVC Via infusionale in PVC plasticizzato con DHEP (verificare compatibilità con farmaci)
 Guscio esterno protettivo in. morbido PVC. Via infusionale in PVC plasticizzato con DHEP. (verificare compatibilità con farmaci)")
31
Membrane elastomeriche
Easypump Membrane elastomeriche INTERNA: Miscela di polimeri ESTERNA: Lattice Il farmaco non viene mai a contatto col lattice Via infusionale In PVC plasticizzato con DHEP incompatibilità con Taxol

32
Descrizione Valvola di calibrazione del flusso Determina ML/H

33
Descrizione La valvola di calibrazione deve essere sempre fissata
alla cute (con cerotto) a garanzia della precisione della portata di infusione FILTRO VALVOLA CALIBRAZIONE
 a garanzia. della precisione della portata. di infusione. FILTRO. VALVOLA CALIBRAZIONE.")
34
Descrizione Connessione LUER-LOCK Valvola unidirezionale
Impossibilità di rimuovere il farmaco dopo il caricamento Codice colore e indicazione di volume (flusso di perfusione in ml/h)
")
35
ST: Short Term: ½ h - 4 h LT: Long Term: 1 - 11 gg. Easypump
LT ml/h 60 ml LT ml/h 100 ml LT ml/h 100 ml LT ml/h 200 ml

36
ISTRUZIONI PER L’USO PRECAUZIONI (…) Esclusivamente monouso. Non riutilizzare, non risterilizzare, nè riempire nuovamente l’unità. Conservare al riparo dalla luce a T ambiente (10- 20°C), umidità relativa compresa tra il 10 e il 90% I farmaci usati con questo sistema devono essere somministrati in base alle istruzioni fornite dalla casa farmaceutica.
, umidità relativa compresa tra il 10 e il 90% I farmaci usati con questo sistema devono essere somministrati in base alle istruzioni fornite dalla casa farmaceutica.")
37
ISTRUZIONI PER L’USO CONTROINDICAZIONI Non usare per somministrare sangue ed emoderivati, soluzioni per NPT, lipidi o emulsioni lipidiche.

38
ISTRUZIONI PER L’USO ISTRUZIONI PER IL RIEMPIMENTO
Adottare una tecnica asettica 1. … 2. … 3. … 4. … 5. … 6. Applicare all’unità l’etichetta (info paziente e farmaco)
")
40
ISTRUZIONI PER L’USO PRECAUZIONI
Gli effettivi tempi di infusione possono variare a causa dei seguenti fattori Temperatura (capillare a contatto con pelle 31°C, cannula sotto i vestiti; se capillare a T ambiente, tempo somministrazione aumenta del 25%) Viscosità della soluzione (vel di flusso nominale per NaCl 09%; con destrosio 5% aumento t somm del 10%) Tempo di inizio somministrazione: iniziare entro 8 h da riempimento (dopo, possibile aumento t somm del 10%) Volume di riempimento: se inferiore al volume nominale, aumento del flusso; se superiore, riduzione del flusso. (…)
 Viscosità della soluzione (vel di flusso nominale per NaCl 09%; con destrosio 5% aumento t somm del 10%) Tempo di inizio somministrazione: iniziare entro 8 h da riempimento (dopo, possibile aumento t somm del 10%) Volume di riempimento: se inferiore al volume nominale, aumento del flusso; se superiore, riduzione del flusso. (…)")
41
ISTRUZIONI PER L’USO Informazioni sui tempi di somministrazione
LT 60-24 100-48 Flusso nom ml/h 2 0.5 Vol riemp nom ml 60 100 270 Vol riemp max ml 65 125 335 Vol trattenuto max ml 3 8

42
ISTRUZIONI PER L’USO Tempi di somministrazione in dipendenza dal volume di riempimento
LT 60-24 100-48 Tempo approssimativo di somministrazione Volume di riempimento (ml) 12 h 38 18 h 42 24 h 52 65 30 h 60 48 h 100 60 h 125
 12 h h h h h h")
43
Tempi di somministrazione in dipendenza dal volume di riempimento
LT Tempo approssimativo di somministrazione Volume di riempimento (ml) 72 h = 3 d 175 96 h = 4 d 215 120 h = 5 d 250 70 135 h = 5.5 d 270 6 d 290 80 7 d 325 89 8 d (192h) 98 9 d (216h) 106 10 d 113 11 d 120
 72 h = 3 d h = 4 d h = 5 d h = 5.5 d d d d (192h) d (216h) d d")
46
I dispositivi di protezione individuale

47
I dispositivi di protezione individuali (DPI)
Direttiva 89/656/CEE recepita dal D.Lgs. 626/94 (tit. IV criteri di scelta e di utilizzo per lavoratori) Direttiva 89/686/CEE, recepita dal D.Lgs. 475/92(requisiti essenziali di sicurezza per fabbricazione, marcatura CE) Provvedimento 5 agosto Linee guida per la sicurezza e la salute dei lavoratori esposti a CTA in ambiente sanitario.
 Direttiva 89/686/CEE, recepita dal D.Lgs. 475/92(requisiti essenziali di sicurezza per fabbricazione, marcatura CE) Provvedimento 5 agosto Linee guida per la sicurezza e la salute dei lavoratori esposti a CTA in ambiente sanitario.")
48
D.Lgs. 4 dicembre 1992, n.475 Attuazione dir. 89/686/CEE
(…)si intendono per DPI i prodotti che hanno la funzione di salvaguardare la persona che l’indossi o comunque li porti con sé da rischi per la salute e la sicurezza. Esclusioni: DPI forze armate, di autodifesa, per uso privato (ombrelli, stivali, guanti per rigovernare e per il calore,…), caschi motocicli.
si intendono per DPI i prodotti che hanno la funzione di salvaguardare la persona che l’indossi o comunque li porti con sé da rischi per la salute e la sicurezza. Esclusioni: DPI forze armate, di autodifesa, per uso privato (ombrelli, stivali, guanti per rigovernare e per il calore,…), caschi motocicli.")
49
I DPI sono suddivisi in tre categorie
I categoria: DPI di progettazione semplice, salvaguardia da danni fisici di lieve entità; l’utilizzatore deve poterne valutare l’efficacia e percepire, prima di riceverne pregiudizio, la progressiva verificazione di effetti lesivi. III categoria: DPI di progettazione complessa, salvaguardia da rischi di morte o di lesioni gravi e di carattere permanente; l’utilizzatore non ha la possibilità di percepire tempestivamente la verificazione istantanea di effetti lesivi. II categoria: DPI che non rientrano in I e III.

50
Alcuni esempi Appartengono alla I cat. i DPI con funzione di salvaguardare da: Azioni lesive superficiali prodotte da strumenti meccanici; Rischi da contatto con oggetti caldi (T non sup a 50°); Ordinari fenomeni atmosferici nel corso di attività professionali. (…) Rientrano esclusivamente nella III categoria: Gli apparecchi di protezione respiratoria filtranti contro gli aerosol solidi, liquidi o contro gas irritanti(…); I DPI che assicurano una protezione limitata nel tempo contro le aggressioni chimiche (…); I DPI per attività in ambienti con condizioni equivalenti ad una T d’aria non inferiore a 100°C
; Ordinari fenomeni atmosferici nel corso di attività professionali. (…) Rientrano esclusivamente nella III categoria: Gli apparecchi di protezione respiratoria filtranti contro gli aerosol solidi, liquidi o contro gas irritanti(…); I DPI che assicurano una protezione limitata nel tempo contro le aggressioni chimiche (…); I DPI per attività in ambienti con condizioni equivalenti ad una T d’aria non inferiore a 100°C.")
51
Procedure di certificazione CE
prima della commercializzazione dei DPI di qualsiasi categoria, il fabbricante deve preparare la documentazione tecnica di costruzione ed effettuare una dichiarazione di conformità CE, (marcatura CE); Prima di produrre un DPI di II o III cat. deve chiedere il rilascio dell’attestato di certificazione CE (organismi di controllo autorizzati).
; Prima di produrre un DPI di II o III cat. deve chiedere il rilascio dell’attestato di certificazione CE (organismi di controllo autorizzati).")
52
Attestato di certificazione CE
E’ l’atto con il quale un organismo di controllo autorizzato attesta che un modello di DPI è stato realizzato in conformità alle disposizioni del presente decreto. Dichiarazione di conformità CE Il fabbricante o il suo rappresentante stabilito nel territorio comunitario, prima di iniziare la commercializzazione, effettua una dichiarazione con la quale attesta che gli esemplari di DPI prodotti sono conformi alle disposizioni del presente decreto, e appone sul DPI la marcatura CE.

53
Requisiti essenziali di salute e sicurezza Allegato II
Requisiti di carattere generale applicabili a tutti i DPI Requisiti supplementari comuni a diverse categorie o tipi di DPI Requisiti supplementari specifici per i rischi da prevenire

54
Requisiti essenziali di salute e sicurezza Allegato II
1. Requisiti di carattere generale applicabili a tutti i DPI I DPI devono assicurare una protezione adeguata contro i rischi 1.1 Principi di progettazione Ergonomia (proteggere senza ostacolare l’attività) Livelli e classi di protezione (max protezione possibile, gradazione della protezione x vari livelli di uno stesso rischio) 1.2 Innocuità dei DPI (non provocano rischi né disturbo, come materiali, superfici a contatto con operatore; min ostacolo ai movimenti). 1.3 Fattori di comfort e di efficacia Adeguamento dei DPI alla morfologia dell’utilizzatore Leggerezza e solidità di costruzione Compatibilità necessaria tra i DPI destinati ad essere indossati simultaneamente 1.4 Nota informativa del fabbricante
 Livelli e classi di protezione (max protezione possibile, gradazione della protezione x vari livelli di uno stesso rischio) 1.2 Innocuità dei DPI (non provocano rischi né disturbo, come materiali, superfici a contatto con operatore; min ostacolo ai movimenti). 1.3 Fattori di comfort e di efficacia Adeguamento dei DPI alla morfologia dell’utilizzatore Leggerezza e solidità di costruzione Compatibilità necessaria tra i DPI destinati ad essere indossati simultaneamente. 1.4 Nota informativa del fabbricante.")
55
1.4 Nota informativa del fabbricante
Preparata e rilasciata obbligatoriamente dal fabbricante per DPI immessi sul mercato; redatta in modo preciso, comprensibile e almeno nella lingua dello Stato membro destinatario Deve contenere, oltre al nome e all’indirizzo del fabbricante o del suo mandatario nella Comunità,ogni informazione utile concernente: Istruzioni di deposito, impiego, pulizia, manutenzione, revisione e disinfezione (…) Le prestazioni ottenute agli esami tecnici effettuati per verificare i livelli o le classi di protezione dei DPI; Gli accessori utilizzabili con i DPI e le caratteristiche dei pezzi di ricambio appropriati; Le classi di protezione adeguate a diversi livelli di rischio e i corrispondenti limiti di utilizzazione; La data o il termine di scadenza dei DPI o di alcuni dei loro componenti; Il tipo di imballaggio appropriato per il trasporto dei DPI; Il significato della marcatura, se presente; Se del caso, i riferimenti delle direttive applicate; Nome, indirizzo, n° di identificazione degli organismi notificati che intrvengono nella certificazione dei DPI.
 Le prestazioni ottenute agli esami tecnici effettuati per verificare i livelli o le classi di protezione dei DPI; Gli accessori utilizzabili con i DPI e le caratteristiche dei pezzi di ricambio appropriati; Le classi di protezione adeguate a diversi livelli di rischio e i corrispondenti limiti di utilizzazione; La data o il termine di scadenza dei DPI o di alcuni dei loro componenti; Il tipo di imballaggio appropriato per il trasporto dei DPI; Il significato della marcatura, se presente; Se del caso, i riferimenti delle direttive applicate; Nome, indirizzo, n° di identificazione degli organismi notificati che intrvengono nella certificazione dei DPI.")
56
Provvedimento 5 agosto 1999 …4.5 Mezzi protettivi individuali
E’ indispensabile durante la manipolazione di CTA indossare i seguenti mezzi protettivi individuali: Guanti (…) Camici (…) Maschere, cuffie, occhiali protettivi (…)
 Camici (…) Maschere, cuffie, occhiali protettivi (…)")
57
Provvedimento 5 agosto 1999 4.5 Mezzi protettivi individuali
Camice rinforzato (TNT) Camice non rinforzato (TNT) Cuffia Mu in TNT Maschere FFP2S Occhiali con prot. laterale Preparazione(cappa f vert.) X Non necessari e Non necessari Preparazione fuori cappa somministrazione x
 Camice non rinforzato (TNT) Cuffia. Mu in TNT. Maschere FFP2S. Occhiali con prot. laterale. Preparazione(cappa f vert.) X. Non necessari e. Non necessari. Preparazione fuori cappa. somministrazione. x.")
58
Provvedimento 5 agosto 1999 4.6.4 operazioni di manutenzione delle cappe e pulizia dei locali
Sostituzione filtri: tuta mu in TNT con cappuccio, maschera facciale a cartuccia (…), guanti, sovrascarpe mu. Pulizia locali preparazione e trattamento: guanti e calzari mu Pulizia servizi igienici pazienti: guanti, maschera FFP2S, calzari mu.
, guanti, sovrascarpe mu. Pulizia locali preparazione e trattamento: guanti e calzari mu. Pulizia servizi igienici pazienti: guanti, maschera FFP2S, calzari mu.")
59
Linee guida S.I.F.O. in oncologia (II Edizione) DPI per manipolazione e somministrazione di CTA
Camice Cuffia o copricapo Telino Guanti per allestimento Occhiali e dispositivi a visiera DP vie respiratorie Sovrascarpe

60
GUANTI PER ALLESTIMENTO
Sono la principale misura di protezione! DPI, terza categoria (D.Lgs. 475/99). Mu, manica lunga, senza polvere, spessore differenziato, spessore min mm se in lattice. Indossare sopra polsini camice, sempre; cambiare ogni 30’ e dopo contaminazione/rottura; per spandimenti/pulizia g. industriali con spessore sup. a 0.45 mm; in somministrazione possibile usare g. standard (Provv. 5/8/99 non distingue)
. Mu, manica lunga, senza polvere, spessore differenziato, spessore min mm se in lattice. Indossare sopra polsini camice, sempre; cambiare ogni 30’ e dopo contaminazione/rottura; per spandimenti/pulizia g. industriali con spessore sup. a 0.45 mm; in somministrazione possibile usare g. standard (Provv. 5/8/99 non distingue)")
61
GUANTI PER ALLESTIMENTO
Permeabilità dipende da: Tipo di materiale Tipo di spessore Tempo di contatto Materiali vari: lattice, nitrile, PU, neoprene. Nessun tipo di materiale è completamente impermeabile a tutti i citotossici: dauno- e doxorubicina attraversano lattice; mostarde azotate attraversano lattice e PVC; PVC, poco elastico, consigliato solo per methotrexate.

64
CAMICE DPI, terza categoria (D.Lgs. 475/99).
Lungo, allacciatura posteriore, maniche lunghe, polsini elastici/a manicotto, impermeabile davanti e maniche. Materiali vari(PP rivestito PE, PE rivestito Tyvek). NON di stoffa. Indossare sempre. Vestibilità, aereazione, addattabilità (D.Lgs. 626/94)
. NON di stoffa. Indossare sempre. Vestibilità, aereazione, addattabilità (D.Lgs. 626/94)")
66
OCCHIALI E DISPOSITIVI A VISIERA
DPI. Protezione laterale, lenti otticamente neutre. Materiali plastici leggeri, antigraffio, antiappannamento. Uso: Occhiali per pulizia cappa, smaltimento CTA, eliminazione escreti del paziente Visiera in somministrazione. In preparazione sotto cappa, evitabili. Pluriuso, lavare come da istruzioni.

67
Dispositivi di protezione delle VIE RESPIRATORIE
DPI, terza categoria, monouso. Semi-maschera rigida, copre naso e bocca. Facciali filtranti, anche con valvola; Classe FFP2 protezione da vapori e aerosol a base acquosa; Classe FFP3 protezione da particelle solide e liquide, compresi vapori di sostanze a base oleosa. Usare facciali filtranti per pulizia cappa e spandimenti; evitabili per preparazione in cappa. Le mascherine chirurgiche non sono DPI.

69
CUFFIA O COPRICAPO DPI, monouso.
Stretto e chiuso in fronte (elastico o allacciatura), deve garantire totale protezione dei capelli e delle orecchie (Provv. 5/8/99: “ Cuffie mu i TNT devono essere utilizzate per proteggere i capelli da possibili contaminazioni.”) Materiale: preferibilmente lo stesso dei camici. Uso suggerito in TUTTE le operazioni di manipolazione dei CTA, compresa la decontaminazione.
, deve garantire totale protezione dei capelli e delle orecchie (Provv. 5/8/99: Cuffie mu i TNT devono essere utilizzate per proteggere i capelli da possibili contaminazioni. ) Materiale: preferibilmente lo stesso dei camici. Uso suggerito in TUTTE le operazioni di manipolazione dei CTA, compresa la decontaminazione.")
70
SOVRASCARPE DPI, monouso. Materiale: in genere PE.
Indossate entrando nel locale di manipolazione, tolte uscendo dal locale, per evitare diffusione della contaminazione.

71
La vigilanza sui dispositivi medici

73
Vigilanza sui dispositivi medici
Art. 9 e 10 del D.Lgs 46/97, attuazione Direttiva 93/42/CEE sui dm Art. 11 del D.Lgs. 507/92,attuazione Dir. 90/385/CEE sui dm impiantabili attivi Art. 11 del D.Lgs. 332/00, attuazione Dir. 98/79/CE sui dispositivi medico-diagnostici in vitro (IVD) Nota informativa CUD 27 luglio 2004 Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm D. Min.Sal. 15 novembre 2005, approvazione schede segnalazione
 Nota informativa CUD 27 luglio Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm. D. Min.Sal. 15 novembre 2005, approvazione schede segnalazione.")
74
Vigilanza sui dispositivi medici
D. Min.Sal. 15 novembre 2005 “Approvazione dei modelli di schede di segnalazioni o mancati incidenti, che coinvolgono dispositivi e dispositivi medico-diagnostici in vitro.” Modelli schede Procedure Incidente e mancato incidente Reclamo

75
Eventi che debbono essere segnalati al Ministero della Salute
D. Min.Sal. 15 novembre 2005, Allegato 8 INCIDENTE MANCATO INCIDENTE

76
Eventi che debbono essere segnalati al Ministero della Salute
D. Min.Sal. 15 novembre 2005, Allegato 8 INCIDENTE: la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonchè qualsiasi carenza nell’etichettatura o nelle istruzioni per l’uso di un dm, abbiano causato, direttamente o indirettamente, un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore

77
Eventi che debbono essere segnalati al Ministero della Salute
D. Min.Sal. 15 novembre 2005, Allegato 8 INCIDENTI INCIDENTE CON ESITO LETALE: incidente in cui il dm ha determinato o ha contribuito a det. il decesso di paziente o utilizzatore. Per stabilire il nesso di causalità si devono tenere in considerazione vari fattori(rischi potenziali nell’utilizzare il dispositivo, caratteristiche dello stesso, condizioni del paziente, ecc.)e deve sempre essere tenuta in considerazione la valutazione effettuata dal medico e/o altro operatore che ha assistito all’evento. Anche se esiste un semplice sospetto l’incidente deve essere notificato.
e deve sempre essere tenuta in considerazione la valutazione effettuata dal medico e/o altro operatore che ha assistito all’evento. Anche se esiste un semplice sospetto l’incidente deve essere notificato.")
78
Eventi che debbono essere segnalati al Ministero della Salute
D. Min.Sal. 15 novembre 2005, Allegato 8 INCIDENTI INCIDENTE IN CUI IL DM HA DETERMINATO UN GRAVE PEGGIORAMENTO DELLO STATO DI SALUTE DEL PAZIENTE O DELL’UTILIZZATORE Per grave peggioramento dello stato di salute si deve intendere: a) una malattia o lesione con pericolo di vita; b) una menomazione di una funzione del corpo o una lesione di una struttura corporea; c) una condizione che rende necessario un intervento medico o chirurgico per impedire una menomazione di una funzione del corpo o una lesione di una struttura corporea; d) una condizione che causa l’ospedalizzazione o il prolungamanto dell’ospedalizzazione
 una malattia o lesione con pericolo di vita; b) una menomazione di una funzione del corpo o una lesione di una struttura corporea; c) una condizione che rende necessario un intervento medico o chirurgico per impedire una menomazione di una funzione del corpo o una lesione di una struttura corporea; d) una condizione che causa l’ospedalizzazione o il prolungamanto dell’ospedalizzazione.")
79
Eventi che debbono essere segnalati al Ministero della Salute
D. Min.Sal. 15 novembre 2005, Allegato 8 MANCATO INCIDENTE: la condizione in cui qualsiasi disfunzione o deterioramento delle caratteristiche o delle prestazioni, nonchè qualsiasi carenza nella etichettatura o nelle istruzioni per l’uso di un dm, a)avrebbe potuto causare, se il dm fosse stato utilizzato, un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore; b)avrebbe potuto causare, durante la procedura d’uso o a seguito della stessa, se non fosse intervenuto il personale sanitario, un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore.
avrebbe potuto causare, se il dm fosse stato utilizzato, un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore; b)avrebbe potuto causare, durante la procedura d’uso o a seguito della stessa, se non fosse intervenuto il personale sanitario, un grave peggioramento dello stato di salute o la morte del paziente o di un utilizzatore.")
80
Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm Commissione Europea

81
“…disfunzione o deterioramento delle caratteristiche o delle prestazioni…” da intendere come un mancato funzionamento di un dm rispetto alla sua destinazione d’uso, quando usato in accordo alle istruzioni del fabbricante. Questo include problemi causati da effetti biologici imprevisti correlati al dm. Destinazione d’uso significa l’uso al quale il dm è destinato secondo i dati forniti dal fabbricante in etichetta, istruzioni per l’uso, materiale promozionale. Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm- Commissione Europea

82
“… qualsiasi carenza nella etichettatura o nelle istruzioni per l’uso …” (“inaccuracy in the labelling, instructions for use and/or promotional materials”) L’inaccuratezza include omissioni o altre carenze. Un’inaccuratezza nelle istruzioni che causa o potrebbe causare uso errato o scorretta manutenzione dovrebbe essere segnalata. Le omissioni non includono l’assenza di informazioni che dovrebbero generalmente essere conosciute dagli utilizzatori previsti. Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm- Commissione Europea

83
Esempi di incidenti e mancati incidenti da segnalare Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm- Commissione Europea Un paziente muore dopo l’uso di un defibrillatore e vi è un’indicazione di un problema con il defibrillatore. L’incidente deve essere segnalato. Una pompa infusionale si blocca, per un malfunzionamento, e non dà l’appropriato allarme; non c’è danno per il paziente. Da segnalare come “mancato incidente”, poiché in una situazione differente l’evento avrebbe potuto causare un danno. Una pompa infusionale rilascia la dose errata a causa di un’incompatibilità tra con il set infusionale usato. Se la combinazione pompa/set utilizzata era indicata nelle istruzioni della pompa o del set, l’evento deve essere segnalato.

84
Esempi di incidenti e mancati incidenti da segnalare Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm- Commissione Europea Un catetere si frattura durante l’inserimento, senza sospetto di operazioni inappropriate. La frattura si verifica in posizione tale che la parte rotta può essere facilmente rimossa, senza intervento chirurgico. Segnalare come mancato incidente. La prematura revisione di un impianto ortopedico è necessaria a causa di un distacco/allentamento. Sebbene la causa non sia ancora stata determinata, questo incidente deve essere segnalato. Il fabbricante fornisce dettagli insufficienti sui metodi di pulizia di strumenti chirurgici riutilizzabili usati in neurchirurgia, nonostante l’ovvio rischio di trasmissione di CJD. Da segnalare come mancato incidente.

85
Esempi di incidenti e mancati incidenti da non segnalare Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm- Commissione Europea INCIDENTI CAUSATI DALLE CONDIZIONI DEL PAZIENTE Un paziente muore dopo una dialisi. Il paziente soffriva di malattia renale in stadio terminale e muore di insufficienza renale. Precoce revisione di un impianto ortopedico dovuto a sviluppo di osteolisi. Un ortopedico impianta una protesi d’anca e vieta l’esecuzione di sport. Il paziente necessita prematura revisione della protesi, dopo essere andato a fare sci d’acqua. INCIDENTI EVITATI GRAZIE A SISTEMI DI ALLARME DI DM Una pompa infusionale si blocca per un malfunzionamento, ma dà l’allarme previsto e non vi è danno per il paziente.

86
Vigilanza sui dispositivi medici
D. Min.Sal. 15 novembre 2005 Art.2 (...) gli operatori sanitari di strutture pubbliche o private debbono effettuare la segnalazione dell’incidente o del mancato incidente, direttamente o tramite la struttura sanitaria di appartenenza, al Ministero della Salute e , possibilmente, anche al fabbricante o mandatario e/o distributore, con la massima urgenza. (...)In ogni caso la segnalazione deve pervenire al Ministero della Salute, per gli incidenti, entro dieci giorni e, per i mancati incidenti, entro trenta giorni dal giorno in cui si è verificato l’evento.
 gli operatori sanitari di strutture pubbliche o private debbono effettuare la segnalazione dell’incidente o del mancato incidente, direttamente o tramite la struttura sanitaria di appartenenza, al Ministero della Salute e , possibilmente, anche al fabbricante o mandatario e/o distributore, con la massima urgenza. (...)In ogni caso la segnalazione deve pervenire al Ministero della Salute, per gli incidenti, entro dieci giorni e, per i mancati incidenti, entro trenta giorni dal giorno in cui si è verificato l’evento.")
87
Rapporto di incidente o di mancato incidente (con DM) da parte di operatori sanitari al Ministero della Salute D.Min.Sal. 15 novembre 2005, Allegato 1 Dati relativi al luogo dove si è verificato l’episodio Dati relativi al dm Dati relativi all’evento Chi è stato coinvolto Dati sull’utilizzo del dm (procedura) Descrizione incidente Conseguenza incidente Dati del compilatore
 Descrizione incidente. Conseguenza incidente. Dati del compilatore.")
88
“Come principio generale, dovrebbe esserci una predisposizione a segnalare piuttosto che a non segnalare in caso di dubbio sulla necessità di segnalare un incidente”. Linea guida MEDDEV 2,12-1 aprile 2001, sui sistemi di vigilanza sui dm- Commissione Europea

89
Grazie per l’attenzione!

Presentazioni simili