Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Termodinamica chimica :
determinare qual è la forza propulsiva delle trasformazioni chimiche e prevederne la spontaneità. Tiene conto SOLO delle proprietà macroscopiche della materia (P, T, V) senza preoccuparsi della sua struttura atomica e molecolare. Le leggi della Termodinamica sono pertanto sganciate dalle teorie atomiche.
 senza preoccuparsi della sua struttura atomica e molecolare. Le leggi della Termodinamica sono pertanto sganciate dalle teorie atomiche.")
2
OBIETTIVI Prevedere quali processi chimici e fisici sono possibili ed in quali condizioni; -Calcolare quantitativamente le proprietà dello stato di equilibrio risultante quando si sviluppa un processo LIMITAZIONI - Non consente di prevedere la velocità di un processo (cinetica); - Non spiega perchè le sostanze devono avere certi valori delle proprietà macroscopiche.
; - Non spiega perchè le sostanze devono avere certi valori delle proprietà macroscopiche.")
3
Modelli astratti per rappresentare sistemi e processi del mondo reale
- condizioni controllate di laboratorio - impianti di produzione industriale - sistemi viventi

4
Sistemi Sistema: porzione di Universo che costituisce l'oggetto della indagine Ambiente: tutto il resto dell'Universo Esempio Sistema Ambiente Sistema Ambiente Sistema isolato: non può scambiare né materia né energia con l'ambiente Vaso di Dewar isolante Esempio Sistema Vuoto

5
Luce e Calore Pistone Mobile Sistema Calore + – Pila
Sistema chiuso: i confini impediscono lo scambio di materia (non di energia) con l'ambiente Esempi Luce e Calore + – Pistone Mobile Pila Sistema aperto: i confini permettono lo scambio con l'ambiente sia di materia che di energia Vapore Calore Sistema Sistema omogeneo : costituito da UNA SOLA FASE (porzione di materia uniforme in ogni punto sia per composizione chimica che per stato fisico) Sistema eterogeneo : costituito da PIU' FASI
 con l ambiente. Esempi. Luce e. Calore. + – Pistone. Mobile. Pila. Sistema aperto: i confini permettono lo scambio con l ambiente sia di materia. che di energia. Vapore. Calore. Sistema. Sistema omogeneo : costituito da UNA SOLA FASE (porzione di materia. uniforme in ogni punto sia per composizione chimica che per stato fisico) Sistema eterogeneo : costituito da PIU FASI.")
6
La porzione rimanente dell’Universo che può scambiare energia (e materia) con il sistema chiuso (aperto) durante il processo in esame è l’AMBIENTE Fornisce le forze esterne che provocano le variazioni delle proprietà del sistema durante un processo. SISTEMA + AMBIENTE = UNIVERSO

7
Stato termodinamico del sistema :
specifica condizione macroscopica nella quale tutte le sue proprietà macroscopiche (volume, massa, T , P) sono note, definite (conoscendo alcune variabili) e mantenute costanti nel tempo intensive estensive Un processo termodinamico modifica lo stato termodinamico del sistema reversibile irreversibile
 sono note, definite (conoscendo alcune variabili) e mantenute costanti nel tempo. intensive estensive. Un processo termodinamico modifica. lo stato termodinamico del sistema. reversibile irreversibile.")
8
Proprietà dei sistemi Intensive: il loro valore NON DIPENDE DALLA MASSA del sistema TEMPERATURA, PRESSIONE, DENSITA', ... Estensive: il loro valore DIPENDE DALLA MASSA del sistema MASSA, VOLUME, PESO, ENERGIA, ... Variabili di stato: lo stato di un sistema è definito quando si possono assegnare valori definiti a tutte le proprietà fisicamente misurabili del sistema (variabili), ad es. P, T, V, ... Quelle variabili il cui valore dipende solo dallo stato del sistema, ma non dal particolare modo in cui si è giunti a tale stato, si dicono variabili o funzioni di stato.
, ad es. P, T, V, ... Quelle variabili. il cui valore dipende solo dallo stato del sistema, ma non dal. particolare modo in cui si è giunti a tale stato, si dicono. variabili o funzioni di stato.")
9
quando nessuna delle proprietà cambia nel tempo,
Dopo che il sistema è stato preparato stabilendo un insieme di vincoli nell’ambiente (ad es. T o P costanti), quando nessuna delle proprietà cambia nel tempo, il sistema ha raggiunto l’equilibrio.
, quando nessuna delle proprietà cambia nel tempo, il sistema ha raggiunto l’equilibrio.")
10
Termodinamica: scienza delle relazioni tra calore e lavoro
termochimica: studia la quantità di calore assorbito o sviluppato nelle reazioni chimiche ad es. combustione di un carburante Energia: proprietà della materia potenziale (capacità) di muoversi esiste in forme diverse che possono convertire una nell’altra : termica, luminosa, elettrica, cinetica,…
 di muoversi. esiste in forme diverse che possono convertire una nell’altra : termica, luminosa, elettrica, cinetica,…")
11
caloria=unità di misura dell’Energia (non-SI) usata dai chimici
Energia cinetica: associata ad un oggetto in virtù del suo moto ½ mv2 = Kg m2/s2 = Joule unità estremamente piccola (P=L/t; watt = 1 J/s watt h = 3600J; 1 Kwatt h= 3.6 x 106 J caloria=unità di misura dell’Energia (non-SI) usata dai chimici quantità di calore (energia) richiesta per innalzare la T di 1 g di H2O di 1°C da 14.5°C a 15.5 °C 1 cal = J Energia potenziale: associata ad un oggetto in virtù della sua posizione in un campo di forze
 usata dai chimici. quantità di calore (energia) richiesta per innalzare la T di 1 g di H2O di 1°C da 14.5°C a 15.5 °C. 1 cal = J. Energia potenziale: associata ad un oggetto in virtù della sua posizione in un campo di forze.")
12
Lavoro : si compie lavoro ogni qualvolta un oggetto viene mosso/spostato contro una forza contraria L = F x l erg = 1 dine x 1cm joule = 1 N x 1 m 1 J = 107 erg Eseguendo lavoro su un sistema varia l’Energia del sistema. Il lavoro meccanico causa ΔEcin o ΔEpot Lavoro meccanico di espansione : un gas si espande in un cilindro dotato di stantuffo mobile passando da Vi a Vf contro una pressione esterna P : L = F × Δh = P × A × Δh = P × ΔV

13
L = P × ΔV = Pascal × m3 = J 1 L atm = 101,325 J

14
Energia interna U: contenuto di energia totale
di un sistema. In un sistema chimico è la somma di vari contributi: - energia potenziale intermolecolare energia cinetica molecolare energia chimica immagazzinata dai legami chimici U : è associata alle forze attrattive e repulsive fra tutti i nuclei e gli elettroni nel sistema -può essere cambiata solo variando lo stato del sistema, ad es. scambiando Lavoro meccanico. -non è possibile determinarne il valore assoluto, ma solo le variazioni (ΔU).
.")
15
Calore o energia termica :
energia che passa in un sistema o fluisce dal sistema in seguito ad una differenza di T tra il sistema termodinamico e l’ambiente. Come il lavoro, anche il calore rappresenta una modalità con la quale è scambiata energia tra un sistema ed il suo ambiente.

16
Il calore fluisce da una regione a T1 maggiore (molecole più veloci) ad una a T2 minore (molecole più lente). Finchè un sistema ed il suo ambiente sono in contatto termico (cioè non termicamente isolati) si ha scambio di energia (calore) tra essi per mantenere la loro T uguale (equilibrio termico). Quando T1 = T2 (molecole a stessa vm), il flusso di calore si arresta.
 si ha scambio di energia (calore) tra essi. per mantenere la loro T uguale (equilibrio termico). Quando T1 = T2 (molecole a stessa vm), il flusso di calore si arresta.")
17
per misurare la quantità di energia trasferita come calore.
Calorimetro : per misurare la quantità di energia trasferita come calore. Se una quantità di calore è trasferita dal sistema al bagno, una parte del ghiaccio fonde ed il volume della miscela acqua e ghiaccio diminuisce. Dalla variazione di volume del contenuto del calorimetro si determina la quantità di calore trasferito.

18
L’equivalenza di calore e lavoro come mezzi per il trasferimento dell’Energia fu suggerita da Benjamin Thompson (1798), consigliere militare del re di Baviera. Notò che : - la quantità di calore prodotto nel perforare i cannoni era proporzionale alla quantità di lavoro eseguito: - il calore non è un “fluido” contenuto nel metallo, perchè l’operazione poteva andare avanti all’infinito. J. Mayer e J. Joule dimostrarono (dal 1840) che la T di una sostanza può essere innalzata sia compiendo su di essa lavoro sia fornendo calore.
 che la T di una sostanza può essere innalzata sia compiendo su di essa lavoro sia fornendo calore.")
19
Una pala mossa dalla caduta di un peso agita l’acqua in un serbatoio.
Così viene eseguito lavoro sull’acqua e la sua T aumenta. L’esperimento è effettuato in un contenitore coibentato in modo da impedire trasferimento di calore tra sistema ed ambiente. La caloria quindi non va più considerata come un’inità indipendente e viene definita come: 1 cal = J

20
Primo principio della termodinamica: ΔU = Q + L
Calore e Lavoro sono energia in transito: - se ΔE è causata da contatto meccanico del sistema con l’ambiente, è compiuto LAVORO; se è causata da un contatto termico, è trasferito CALORE (finchè non si realizza l’uguaglianza delle T). Spesso sia il LAVORO che il CALORE attraversano i confini del sistema e la variazione di energia interna (ΔU) è la somma dei due contributi. Primo principio della termodinamica: ΔU = Q + L
. Spesso sia il LAVORO che il CALORE attraversano i confini del sistema e la variazione di energia interna (ΔU) è la somma dei due contributi. Primo principio della termodinamica: ΔU = Q + L.")
21
Q e L dipendono dal processo (percorso) che unisce due stati,
ma la loro somma ΔU è indipendente dal percorso, quindi U è una funzione di stato. In qualunque processo, il calore ceduto al sistema è rimosso dall’ambiente : Q sis = - Qamb Allo stesso modo, il lavoro compiuto sul sistema è fatto dall’ambiente : Lsis = - Lamb Sommando le due relazioni si ottiene: ΔUsis = - ΔUamb

22
Pertanto, le variazioni di energia del sistema e dell’ambiente hanno lo stesso valore, ma segno opposto. La variazione dell’energia totale dell’universo (sistema + ambiente) è quindi: ΔUuniv = ΔUsis + ΔUamb In qualunque processo l’energia totale dell’universo rimane invariata; l’energia totale si conserva mentre è scambiata tra sistema ed ambiente.
 è quindi: ΔUuniv = ΔUsis + ΔUamb. In qualunque processo l’energia totale dell’universo rimane invariata; l’energia totale si conserva mentre è scambiata tra sistema ed ambiente.")
23
Il lavoro non è una funzione di stato :
- se il processo è effettuato in modo repentino il lavoro è < se il lavoro è effettuato in modo reversibile, il lavoro è notevolmente > una trasformazione reversibile fornisce il max lavoro ottenibile. si può tornare allo stato iniziale senza che né il sistema né l’ambiente subiscano modifiche permanenti di entità apprezzabile si procede mediante una serie infinita di stati, in ciascuno dei quali il sistema è in equilibrio per realizzare una trasformazione reversibile occorre un tempo infinito

24
Processo reversibile:
procede in modo che i valori delle sue funzioni di stato non differiscano più di un infinitesimo tra due istanti successivi della trasformazione (ideale). Lrev > Lirrev Processo irreversibile: procede in modo spontaneo e i valori delle sue funzioni di stato differiscono di quantità finite da un istante al successivo della trasformazione (naturale). Qrev > Qirrev
. Lrev > Lirrev. Processo irreversibile: procede in modo spontaneo e i valori delle sue funzioni di stato differiscono di quantità finite da un istante al successivo della trasformazione (naturale). Qrev > Qirrev.")
25
Convenzione dei segni Calore Calore + Q - Q Sistema Lavoro Lavoro - L

26
Forme diverse di energia
Energia cinetica v E = ½ mv2 Energia potenziale: di gravità (a) (b)
 (b)")
27
Energia potenziale: elastica (a) (b) chimica TNT (a) (b)
 (b) chimica TNT (a) (b)")
28
Alcuni fattori di conversione
tra unità energetiche 1 Erg = 10-7 J ; 1 litro x atmosfera = 101,3 J 1 eV = 1,602 • J ; 1 caloria = 4,184 J

29
Alcuni tipi importanti di trasformazioni
CICLICHE: se il sistema torna allo stato iniziale; ISOTERME: se avvengono a temperatura costante; ISOBARE: se avvengono a pressione costante; ISOCORE: se avvengono a volume costante; ADIABATICHE: se avvengono senza scambio di calore.

30
L'Energia NON si crea NE' si distrugge, ma si TRASFORMA
Primo Principio della Termodinamica I° ENUNCIATO: L'Energia NON si crea NE' si distrugge, ma si TRASFORMA h Esperienza di Joule: equivalente meccanico della caloria Equivalente elettrico della caloria 1 cal = 4,184 J

31
2° Enunciato: L'energia interna U di un sistema è una
funzione di stato Scambia calore e lavoro con l'ambiente Sistema Chiuso Stato I Sistema Chiuso Stato II U2 = U1 + Q - L ; U2 - U1 = Q - L ; DU = Q -L Processi ciclici Stato I UFINALE = UINIZIALE ; DU = O ; Q = L U è una variabile estensiva, di cui si possono misurare SOLO le variazioni

32
I° principio della termodinamica: principio di equivalenza
la scomparsa di un certo tipo di energia deve essere accompagnata dalla comparsa di una equivalente quantità di una forma diversa di energia pertanto le varie forme di energia si trasformano una nell’altra secondo rapporti fissi che vengono valutati mediante le trasformazioni cicliche (stato iniziale e finale coincidono) ΔU = Q – L = Q = L
 ΔU = Q – L = 0 Q = L.")
33
H = U + PV Processi adiabatici (Q = O) DU = - L
Processi isocori (L = O) DU = Qv Processi isobari, con solo lavoro di espansione L = + PDV DU = Qp - PDV ; Qp = DU + PDV H = U + PV ENTALPIA Per una trasformazione isobara DH = DU + PDV DH = Qp H è una funzione di stato come U, e come per U se ne misurano le variazione DH, ma non il valore assoluto, che non è determinabile. H è variabile ESTENSIVA.
 DU = Qv. Processi isobari, con solo lavoro. di espansione L = + PDV. DU = Qp - PDV ; Qp = DU + PDV. H = U + PV. ENTALPIA. Per una trasformazione isobara DH = DU + PDV. DH = Qp. H è una funzione di stato come U, e come per U se ne misurano le. variazione DH, ma non il valore assoluto, che non è determinabile. H è variabile ESTENSIVA.")
34
L’energia interna e l’entalpia sono grandezze simmetriche
che si utilizzano in caso di volume costante ΔU = Qv o pressione costante ΔH = Qp

35
Si valuti la differenza tra D H e D U per una reazione :
poiché D H = D U + D(PV) la differenza è racchiusa nel termine D(PV). Nelle reazioni a cui partecipano solidi e liquidi, DV sono trascurabili, per cui D (P V) = e D H = D U Per le reazioni tra gas, P V = n R T da cui D(P V) = (D n) R e quindi D H = D U + (D n) R T Nelle reazioni tra gas vi è differenza tra DH e DU solo quando nel passaggio da reagenti a prodotti si ha Dn diverso da 0.
 la differenza è racchiusa nel termine D(PV). Nelle reazioni a cui partecipano solidi e liquidi, DV sono trascurabili, per cui D (P V) = 0 e D H = D U. Per le reazioni tra gas, P V = n R T. da cui D(P V) = (D n) R. e quindi D H = D U + (D n) R T. Nelle reazioni tra gas vi è differenza tra DH e DU solo quando nel passaggio da reagenti a prodotti si ha Dn diverso da 0.")
36
E’ possibile misurare la variazione del contenuto energetico di un sistema a seguito di una trasformazione, ossia il DH, attraverso la quantità di Qp. E’ anche possibile calcolare teoricamente tale grandezza e anziché conoscere il ΔH di una reazione chimica in particolari condizioni, conviene conoscere il ΔH°, ossia la variazione di entalpia in condizioni standard (o normale).
.")
37
La funzione di stato entalpia esprime la quantità di energia che
un sistema può scambiare con l’ambiente. Dipende da: - la MASSA di materia del sistema CONDIZIONI FISICHE STATO di AGGREGAZIONE - POLIMORFISMO condizioni standard o di riferimento : - 1 mole 25 °C ; 1 atm gas, liquido, solido forma cristallina stabile nelle condizioni std (ad es. grafite)
")
38
TONALITA’ TERMICA : calore liberato o assorbito nel corso di una reazione chimica (spesso P =cost → Qp =ΔH l’entalpia è l’energia trasferita come calore a P cost) reazioni ESOTERMICHE : calore liberato (–Qp) ΔH <0 reazioni ENDOTERMICHE : calore assorbito (+Qp) ΔH >0 reazioni TERMONEUTRALI : effetti termici nulli (Qp=ΔH=0)
 reazioni ESOTERMICHE : calore liberato (–Qp) ΔH <0. reazioni ENDOTERMICHE : calore assorbito (+Qp) ΔH >0. reazioni TERMONEUTRALI : effetti termici nulli (Qp=ΔH=0)")
39
Agitatore Termometro Contenitore isolante Contenitore metallico Acqua Ossigeno gassoso ad alta pressione Bomba d'acciaio Sostanza da bruciare

40
Entalpie di formazione dei composti, ΔH°f : entalpia associata alla sintesi di 1 mole di composto a partire dagli elementi nei relativi stati std. Quelle degli elementi sono nulle per convenzione. ΔHstd della reazione di combustione di 1 mole di composto in eccesso di O2 gas

41
La tonalità termica (quantità di calore sviluppato o assorbito a P cost) di una reazione chimica dipende solo dallo stato iniziale e finale dei reagenti e dei prodotti, mentre è indipendente dal numero dei processi intermedi e dall’ordine con cui sono effettuati. Reattivi Stato intermedio Prodotti D H 1 H' 2 H'' = + C (s) O 2(g) CO (g) H° 3 Stato Finale Stato Iniziale + O + O CO + A + B C + D Secondo la legge di Hess : r = (H° fC + H° fD ) - (H° fA fB Elementi Componenti Prodotti C + D Reattivi A + B (Diretta) I = H° II deve essere : ;
 di una reazione chimica dipende solo dallo stato iniziale e finale dei reagenti e dei prodotti, mentre è indipendente dal numero dei processi intermedi e dall’ordine con cui sono effettuati. Reattivi. Stato intermedio. Prodotti. D. H. 1. H 2. H = + C. (s) O. 2(g) CO. (g) ½. H° 3. Stato Finale. Stato Iniziale. + O. + O. CO + A + B. C + D. Secondo la legge di. Hess. : r. = (H° fC. + H° fD. ) - (H° fA. fB. Elementi Componenti. Prodotti C + D. Reattivi A + B. (Diretta) I. = H° II. deve essere : ;")
42
Pertanto, l’evoluzione del calore (ΔH) non è un criterio
Le trasformazioni spontanee avvengono senza intervento esterno e solo nella direzione che porta al raggiungimento dell’equilibrio. Non conosciamo la velocità né le condizioni di reazione necessarie per raggiungere l’equilibrio (cinetica). - Molti processi spontanei sono esotermici (ΔH<0) : ad es. acido + base → sale + acqua (neutralizzazione); ma numerosi processi spontanei sono endotermici (ΔH>0) : ad es dissoluzione di NH4NO3 in acqua; - espansione di un gas nel vuoto; - fusione del ghiaccio Pertanto, l’evoluzione del calore (ΔH) non è un criterio sufficiente a determinare la spontaneità di un processo.
. - Molti processi spontanei sono esotermici (ΔH<0) : ad es. acido + base → sale + acqua (neutralizzazione); ma numerosi processi spontanei sono endotermici (ΔH>0) : ad es. - dissoluzione di NH4NO3 in acqua; - espansione di un gas nel vuoto; - fusione del ghiaccio. Pertanto, l’evoluzione del calore (ΔH) non è un criterio. sufficiente a determinare la spontaneità di un processo.")
44
In un processo spontaneo,
l’energia va in direzione della massima dispersione (es. consideriamo un libro ad una certa altezza dal pavimento: all’inizio l’energia è tutta concentrata nel libro come Epot; dopo la caduta del grave, l’Etot è la stessa ma è dispersa nell’aria, sul pavimento, nel libro.) La funzione di stato entropia, S, ci consente di quantizzare questa dispersione di energia. L’energia potenziale viene dispersa quando è convertita in energia termica. La dispersione avviene quando l’energia è trasferita sotto forma di calore (q). L’effetto di una certa quantità di energia trasferita come calore (q) sulla dispersione dipende dalla T (effetto > a basse T) e dal tipo di processo (reversibile o irreversibile)
 La funzione di stato entropia, S, ci consente di quantizzare. questa dispersione di energia. L’energia potenziale viene dispersa quando è convertita in energia termica. La dispersione avviene quando l’energia è trasferita sotto forma di calore (q). L’effetto di una certa quantità di energia trasferita come calore (q) sulla dispersione dipende dalla T (effetto > a basse T) e dal tipo di processo (reversibile o irreversibile)")
45
Entropia S ha la dimensioni di una energia/°K e si misura in Qrev Qirr
DS = (Rev) DS > (Irr) T T S ha la dimensioni di una energia/°K e si misura in cal/grado (Unità Entropiche, UE) o Joule/ °K. E' una variabile estensiva come U e H.
 DS > (Irr) T. T. S ha la dimensioni di una energia/°K e si misura in. cal/grado (Unità Entropiche, UE) o Joule/ °K. E una variabile estensiva come U e H.")
46
"S è una funzione di Stato"
Secondo Principio della Termodinamica "S è una funzione di Stato" In un sistema isolato (es. l'Universo) sede di trasformazioni irrev Q Q = O DS > DS > O T "L'entropia dell'Universo tende ad un valore massimo“ Se un sistema isolato è in equilibrio, cioè sede di trasformazioni rev dQ Q = O dS = dQ = TdS =0 (Equilibrio) T "L'entropia dell'Universo è costante“
 sede di trasformazioni irrev. Q. Q = O. DS > DS > O. T. L entropia dell Universo tende ad un valore massimo Se un sistema isolato è in equilibrio, cioè sede di trasformazioni rev. dQ. Q = O. dS = dQ = TdS =0. (Equilibrio) T. L entropia dell Universo è costante")
47
(trasf. inversa spontanea) T
Se il sistema è sede di trasformazioni irreversibili (spontanee) dQ dS > dQ < TdS T Se invece si ha, per una ipotetica trasformazione dQ dS < (trasf. inversa spontanea) T Tale trasformazione non è spontanea, ma anzi è spontanea quella inversa. S è definita in modo tale che il segno algebrico della sua variazione, considerando il valore totale di sistema + ambiente, risulti positivo nella direzione del processo spontaneo.
 dQ. dS > dQ < TdS. T. Se invece si ha, per una ipotetica trasformazione. dQ. dS < (trasf. inversa spontanea) T. Tale trasformazione non è spontanea, ma anzi è spontanea quella inversa. S è definita in modo tale che il segno algebrico della sua variazione, considerando il valore totale di sistema + ambiente, risulti positivo nella direzione del processo spontaneo.")
48
La variazione di entropia ΔS si calcola come visto nel caso del ΔH:
ΔS = Sf – Si Ad es. per la fusione del ghiaccio H2O(s) → H2O(l) ΔS = ( ) J/K = 22 J/K nel passaggio di stato Solido → Liquido : S aumenta
 → H2O(l) ΔS = ( ) J/K = 22 J/K. nel passaggio di stato Solido → Liquido : S aumenta.")
49
Per sistemi isolati (Trasf. Adiabatiche)
dQ = 0 (trasformazione adiabatica) dQ dS = dS = 0 EQUILIBRIO T dQ dS > dS > 0 SPONTANEITA' T dQ dS < dS < 0 TRASF. INVERSA SPONTANEA T
 dQ. dS = dS = 0 EQUILIBRIO. T. dQ. dS > dS > 0 SPONTANEITA T. dQ. dS < dS < 0 TRASF. INVERSA SPONTANEA. T.")
50
Processo spontaneo (irreversibile): - può avvenire da solo, senza
Processo spontaneo (irreversibile): - può avvenire da solo, senza intervento esterno; - segue una direzione definita. Il I° Principio non spiega la direzionalità dei processi spontanei: l’energia è conservata sia nel processo diretto che inverso e nulla indica una preferenza per una direzione o l’altra.
: - può avvenire da solo, senza intervento esterno; - segue una direzione definita. Il I° Principio non spiega la direzionalità dei processi spontanei: l’energia è conservata sia nel processo diretto che inverso e. nulla indica una preferenza per una direzione o l’altra.")
51
PROCESSI FISICI SPONTANEI :
. Il calore fluisce da un corpo caldo ad uno freddo (mai il contrario). . Un gas espande in una regione a pressione più bassa. . Un soluto si dissolve in un solvente per formare un sistema omogeneo. PROCESSI CHIMICI SPONTANEI : . una miscela di H2 e O2 brucia (se “accesa”) in modo esplosivo per dare H2O; non si osserverà mai la decomposizione spontanea di H2O in H2 e O2; . 2 Na + Cl2→ 2 NaCl (spontanea) . 2 Cu + O2 → 2 CuO (spontanea)
. . Un gas espande in una regione a pressione più bassa. . Un soluto si dissolve in un solvente per formare un sistema omogeneo. PROCESSI CHIMICI SPONTANEI : . una miscela di H2 e O2 brucia (se accesa ) in modo esplosivo per dare H2O; non si osserverà mai la decomposizione spontanea di H2O in H2 e O2; . 2 Na + Cl2→ 2 NaCl (spontanea) . 2 Cu + O2 → 2 CuO (spontanea)")
52
Entropia ed Energia sono ben diverse:
L’energia non si crea né si distrugge. L’entropia si crea/aumenta durante qualsiasi processo spontaneo. Quando un flusso di calore entra o esce dal sistema, l’entropia accompagna questo flusso poichè l’energia termica è dovuta al movimento casuale delle particelle (dispersione). Il calore tende a rendere più disordinato il sistema verso cui fluisce. L. Boltzmann ( ) ha correlato S con W (n° possibili diverse disposizioni delle molecole in un sistema; ogni disposizione ha stessa energia)
. Il calore tende a rendere più disordinato il sistema verso cui. fluisce. L. Boltzmann ( ) ha correlato S con W. (n° possibili diverse disposizioni delle molecole in un sistema; ogni disposizione ha stessa energia)")
53
Lo stato termodinamico di un sistema può essere definito in due modi:
descrizione macroscopica (P, T, V,…) descrizione microscopica (proprietà delle singole particelle) Devono coincidere poiché la prima riporta proprietà medie di tutti gli stati microscopici del sistema. Usando la teoria della probabilità possiamo predire e spiegare il comportamento dell’insieme molto numeroso di atomi e molecole che compongono un campione macroscopico di materia.
 descrizione microscopica (proprietà delle singole particelle) Devono coincidere poiché la prima riporta proprietà medie di tutti gli. stati microscopici del sistema. Usando la teoria della probabilità possiamo predire e spiegare il comportamento dell’insieme molto numeroso di atomi e molecole che compongono un campione macroscopico di materia.")
54
Espansione libera adiabatica di una mole di gas ideale nel vuoto :
processo spontaneo. La probabilità che la molecola si trovi a sinistra sarà uguale a ½. Direzione di un processo spontaneo: confronto tra la probabilità di trovare tutte le molecole nel lato sinistro con quella di trovare le molecole uniformemente distribuite sui due lati.

55
( ) 1 2 talmente bassa da poterla considerare nulla.
In un gas formato da 4 molecole, la probabilità che si trovino tutte a sinistra è : 1 × 1 × 1 × 1 = 1 Per N molecole, la probabilità è 6.023×1023 ( 1 2 ) talmente bassa da poterla considerare nulla. Quindi l’interpretazione statistica molecolare spiega che un gas si espande per riempire tutto il volume disponibile perchè questa configurazione è molto più probabile di quella in cui tutte le molecole si trovano nel lato sinistro.
 talmente bassa da poterla considerare nulla. Quindi l’interpretazione statistica molecolare spiega che un gas si espande per riempire tutto il volume disponibile perchè questa configurazione è molto più probabile di quella in cui tutte le molecole si trovano nel lato sinistro.")
56
Nulla impedisce la compressione spontanea di un gas,
ma l’evento è talmente improbabile da non essere mai stato osservato. La spontaneità in Natura deriva dal comportamento casuale e statistico di un enorme numero di molecole. La direzionalità di un cambiamento spontaneo è una conseguenza delle tantissime molecole presenti nei sistemi macroscopici trattati dalla termodinamica.

57
Funzione di stato entropia S : aumenta quando il processo è spontaneo.
Rimuovendo i vincoli, le molecole si muovono per esplorare il range dei moti possibili, che ora è aumentato. Si trova il numero di stati microscopici (microstati, W) disponibili per le molecole : tiene conto di tutte le possibili combinazioni delle posizioni e delle quantità di moto disponibili per le N molecole.
 disponibili per le molecole : tiene conto di tutte le possibili combinazioni delle posizioni e delle quantità di moto disponibili per le N molecole.")
58
Interpretazione microscopica (TERMODINAMICA STATISTICA):
l’entropia è una misura del numero di stati microscopici con cui si può descrivere un determinato stato macroscopico S = k ln W W= numero dei microstati disponibili alle molecole; k = R / N , costante di Boltzmann (1.38 × J/K) Quando un sistema aumenta il suo disordine significa che è aumentato il n° di possibili disposizioni molecolari per quel sistema. Un aumento del caos molecolare comporta un aumento di entropia.
 Quando un sistema aumenta il suo disordine significa che è aumentato il n° di possibili disposizioni molecolari per quel sistema. Un aumento del caos molecolare comporta un aumento di entropia.")
60
I seguenti processi sono accompagnati da aumento di S :
1 I seguenti processi sono accompagnati da aumento di S : . frammentazione di una molecola grande; . aumento del numero di moli di gas; . fusione di un solido; . evaporazione di un liquido; . mescolamento di due sostanze; . discoglimento di una sostanza nell’altra. I seguenti processi sono accompagnati da diminuzione di S : . associazione di piccole molecole in molecole più grandi; . condensazione di un gas; . reazione di un gas per formare un solido o un liquido; . congelamento di un liquido; . cristallizzazione di una sostanza disciolta in un solvente; . reazioni in soluzione per formare un precipitato.

61
Il fatto che in alcuni processi S diminuisce non è in contrasto con il II° Principio della Termodinamica : a differenza dell’Universo (dove S è costante o aumenta), in un sistema S può diminuire. Ciò implica che nell’ambiente S debba crescere in misura uguale (processo rev) o maggiore (processo irrev).
, in un sistema S può diminuire. Ciò implica che nell’ambiente S debba crescere in misura uguale (processo rev) o maggiore (processo irrev).")
62
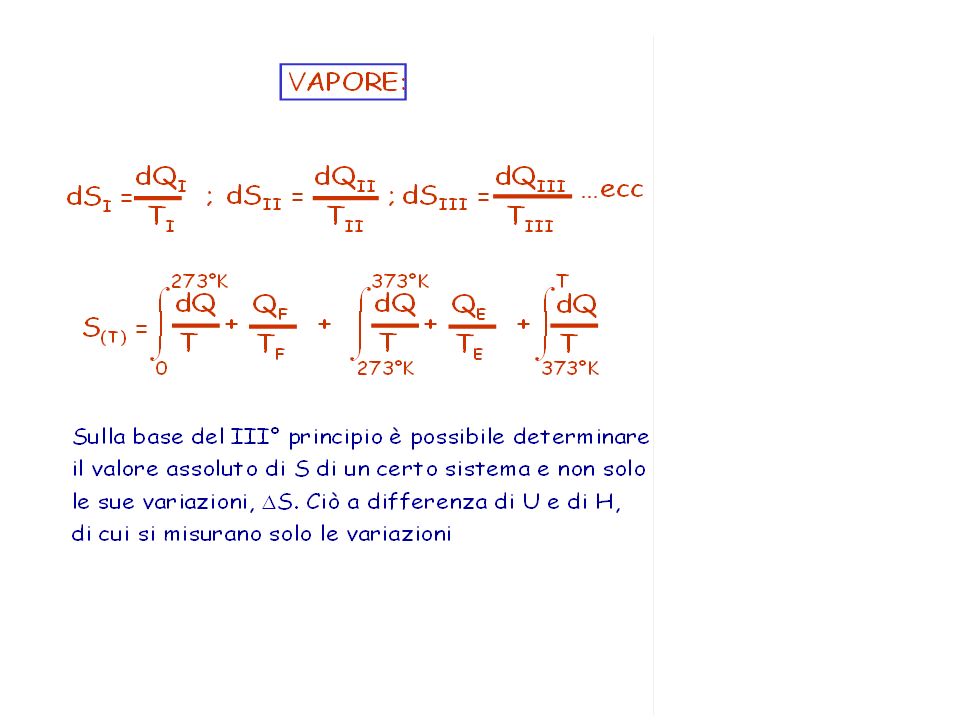
Allo zero assoluto (0 K) ciascuna particella di un solido cristallino puro e perfetto occupa posizioni precise e fisse nel reticolo cristallino. Poichè è assente qualsiasi tipo di disordine ed i moti termici si annullano, il numero di microstati disponibili ad una sostanza a 0 K è pari ad 1 (uno) (W = 1, poichè esiste solo una disposizione possibile per le molecole) e l’entropia assoluta si annulla S = k ln 1 = 0 E’ possibile quindi conoscere il valore assoluto dell’entropia.
 (W = 1, poichè esiste solo una disposizione possibile per le molecole) e l’entropia assoluta si annulla. S = k ln 1 = 0. E’ possibile quindi conoscere il valore assoluto dell’entropia.")
63
a 25°C e 1 atm si ottiene S° (entropia assoluta normale) = 1438/273 = 5.27 UE/mole
 = 1438/273 = 5.27 UE/mole")
65
Una molecola che si spezza in due molecole più piccole.
Aumento di moli gassose.

66
ΔS° = S°(prodotti) - S°(reagenti)
Calcolare la variazione di entropia std ΔS° per le seguenti reazione : CaCO3(s) → CaO (s) + CO2(g) S°= 92.9 J mol-1 K-1 S°= J mol-1 K-1 S°= J mol-1 K-1 ΔS° = ( ) – 92.9 = J mol-1 K-1
 → CaO (s) + CO2(g) S°= 92.9 J mol-1 K-1. S°= J mol-1 K-1. S°= J mol-1 K-1. ΔS° = ( ) – 92.9 = J mol-1 K-1.")
67
L’entropia è la prima funzione termodinamica in grado di fornire indicazioni sulla spontaneità di una trasformazione chimica. Il II° Principio della Termodinamica implica che : per un processo rev (non spontaneo), Δ S=0 per un processo irrev (spontaneo), Δ S>0 Tuttavia qui si fa riferimento all’Universo, mentre noi siamo interessati al sistema dove avviene la reazione chimica.
, Δ S=0. per un processo irrev (spontaneo), Δ S>0. Tuttavia. qui si fa riferimento all’Universo, mentre noi siamo interessati al sistema dove avviene la reazione chimica.")
69
H = G + TS Di norma le reazioni avvengono a T e P costanti
a P cost il calore scambiato è ΔH = Qp a T cost il calore scambiato non può venire totalmente trasformato in altre forme di energia. Il calore può essere scomposto in due parti: . una parte libera di trasformarsi in altre forme di energia; . un’altra parte necessaria a mantenere il sistema alla temperatura stabilita e quindi non utilizzabile per la conversione in altre forme di energia. entalpia = energia libera + energia inutilizzabile H = G + TS

70
L’entalpia H di un sistema è tutta energia utilizzabile (H = G)
solo a T = 0 K quando S = 0

73
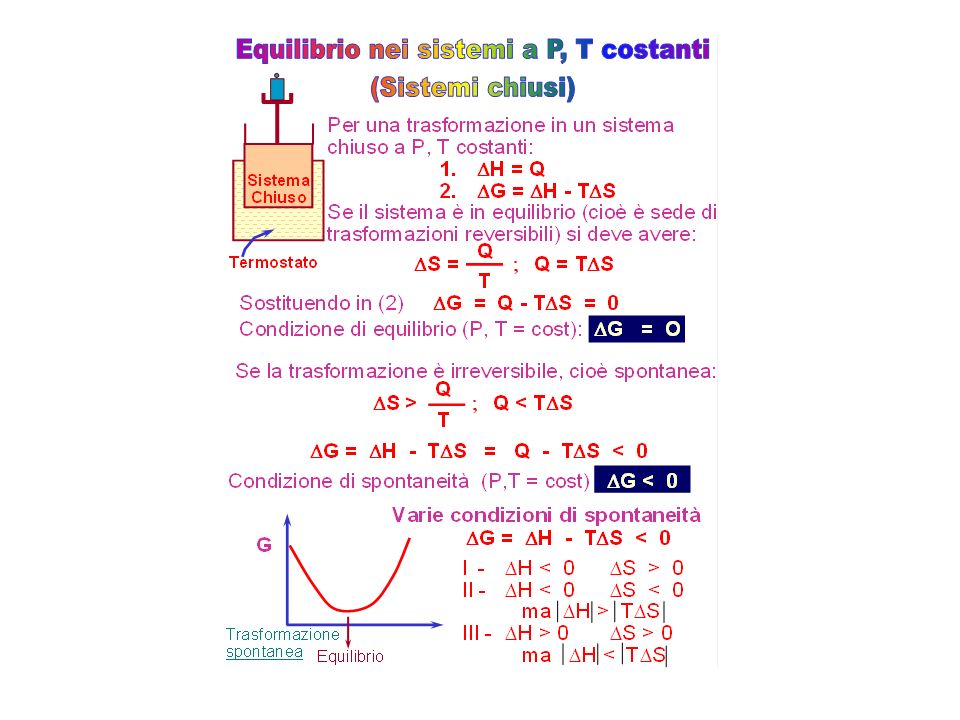
Criteri termodinamici di equilibrio e di spontaneità di un processo
II II Q Acqua U Ghiaccio Q 1 mole ghiaccio 1 mole acqua liquida Spontanea a t > O° Fusione: processo endotermico Ghiaccio Acqua Spontanea a t < O° Occorre considerare DS insieme a DH Fattore Energetico Fattore Entropico Ogni sistema tende a raggiungere il minimo contenuto di energia Ogni sistema tende a raggiungere il massimo contenuto di entropia Ciò spiega perché nei sistemi isolati (in cui il contenuto di energia deve restare costante) l'unico criterio di spontaneità di un processo si basa sull'aumento di S.
 l unico criterio di spontaneità di un processo si basa sull aumento di S.")
74
Combinazione dei fattori energetico
ed entropico Se un processo avviene con diminuzione di energia e con aumento di entropia, esso è spontaneo I Caso S S DS > O DH < O S H Concordanza dei fattori energetico ed entropico Esempio: Calore H2 + Cl2 HCl II Caso In un sistema isolato (DH=O) una trasformazione è spontanea se DS > 0, cioè se cresce l'entropia S DS > O DH = O Solo fattore entropico Esempio: Dissoluzione dello zucchero in acqua
 una trasformazione è spontanea. se DS > 0, cioè se cresce l entropia. S. DS > O. DH = O. Solo fattore entropico. Esempio: Dissoluzione dello. zucchero in acqua.")
75
DS > O, DH > 0 (a) DS < O, DH < 0 (b) DS DH III Caso
Contrasto tra il fattore energetico e quello entropico DS > O, DH > 0 (a) DS < O, DH < 0 (b) Esempio: (a) FUSIONE del GHIACCIO (b) SOLIDIFICAZIONE DELL'ACQUA DECIDE LA TEMPERATURA TEMPERATURA K DS DH
 DS < O, DH < 0 (b) Esempio: (a) FUSIONE del GHIACCIO. (b) SOLIDIFICAZIONE DELL ACQUA. DECIDE LA. TEMPERATURA. TEMPERATURA. K. DS. DH.")
81
G° è caratteristica della sostanza
(concentrazione ideale)
")
83
SISTEMI AD UN COMPONENTE
EQUILIBRI TRA LE FASI SISTEMI AD UN COMPONENTE singola sostanza pura che può esistere in diversi stati di aggregazione

84
Cambiamenti di Stato LIQUIDO SOLIDO GAS t P = 1 atm + + Q (Calore) QF
Fusione LIQUIDO Evaporazione Solidificazione Condensazione Sublimazione SOLIDO GAS Brinazione Pesi per regolare la pressione Termometro Pistone mobile Cilindro di vetro 1 Mole di acqua Q Q Sorgente calda o fredda t °C P = 1 atm 100° - Solido Vapore + Liquido Solido + Liquido Vapore 0° - Liquido Q (Calore) QF QE
 QF. QE.")
85
Gli equilibri tra le fasi dipendono dalla P :
calore latente di fusione, QF = - calore latente di solidificazione, QSOLID calore latente di evaporazione, QE = - calore latente di condensazione, QC a P = cost, QF = ΔHF QSOLIF = ΔHS Gli equilibri tra le fasi dipendono dalla P : la curva di riscaldamento di H2O a P= 1 torr è diversa da quella a P= 1 atm

86
P = 0,01 torr P = 100 atm (76.000 torr) T (°C) Q (Calorie) QS T (°C) Q
Solido Solido + Vapore Vapore O°C Q (Calorie) QS P = 100 atm ( torr) T (°C) Vapore 312°C Solido + Liquido Liquido Liquido + Vapore Solido O°C Q (Calorie) QF QE
 QS. P = 100 atm ( torr) T. (°C) Vapore. 312°C. Solido. + Liquido. Liquido. Liquido. + Vapore. Solido. O°C. Q. (Calorie) QF. QE.")
87
temperature di equilibrio tra le fasi
DIAGRAMMA DI STATO pressione temperature di equilibrio tra le fasi

88
temperature di equilibrio tra le fasi
curva AO: equilibrio solido-vapore (come varia la P del vapore in equilibrio con il solido al variare della T) curva OB: equilibrio solido-liquido (come varia la T fusione del solido con la P) curva OC: equilibrio liquido-vapore (come varia la T ebollizione del liquido con la P) S L V P T temperature di equilibrio tra le fasi pressione O C B A
 curva OB: equilibrio solido-liquido. (come varia la T fusione del solido con la P) curva OC: equilibrio liquido-vapore. (come varia la T ebollizione del liquido con la P) S. L. V. P. T. temperature di equilibrio tra le fasi. pressione. O. C. B. A.")
89
La curva che descrive l’equilibrio LIQUIDO VAPORE
si interrompe al punto critico (Tc, Pc). In queste condizioni la sostanza è detta FLUIDO SUPERCRITICO è un gas con una P tanto elevata da avere la densità di un liquido; - è un liquido con una viscosità simile a quella di un gas.
. In queste condizioni la sostanza è detta. FLUIDO SUPERCRITICO. è un gas con una P tanto elevata da avere la densità di un liquido; - è un liquido con una viscosità simile a quella di un gas.")
90
E’ possibile determinare l’andamento delle curve di
equilibrio tra le due fasi in base alle relazioni della termodinamica?

91
Equazione di Clausius - Clapeyron
Considerando una mole di liquido in equilibrio con una mole di gas a P, T costanti, deve essere : GL = GV Alterando di un infinitesimo dT la temperatura T e di un infinitesimo dP la pressione P in modo tale che il sistema resti in equilibrio, si avrà: GL + dGL = GV + dGV dGL = dGV dGV = VvdP - SVdT dGL = VLdP - SLdT VVdP - SVdT = VLdP - SLdT (VV - VL)dP = (SV - SL)dT = DSdT QE QE DS = QE = Calore latente di evaporazione T QE dP dT (VV - VL)dP = QE = T dT (VV - VL)T dP QE > 0, perciò > 0 (La tensione di vapore cresce con T) Trascurando VL rispetto a VV, ed assumendo valida la legge dei gas ideali per il vapore: VV = dT RT/P QE QEP dP QE = = = RT . dT VVT T RT2 dP QE dT P QE 1 forma differenziale = ; d (lnP) = d P R T2 R T QE - QE - RT lnP = cost P = cost. e forma integrata RT
dP = (SV - SL)dT = DSdT. QE. QE. DS = QE = Calore latente di evaporazione. T. QE. dP. dT. (VV - VL)dP = QE. = T. dT. (VV - VL)T. dP. QE > 0, perciò > 0 (La tensione di vapore cresce con T) Trascurando VL rispetto a VV, ed assumendo valida la legge dei gas ideali per il vapore: VV = dT. RT/P. QE. QEP. dP. QE. = = = RT. . dT. VVT. T. RT2. dP. QE. dT. P. QE. 1. forma differenziale. = ; d (lnP) = - d. P. R. T2. R. T. QE. - QE. - RT. lnP = + cost P = cost. e. forma integrata. RT.")
92
Equilibrio gas - solido
lnP P Acqua Alcool Etilico Alcool Etilico Acqua 1 T T Equilibrio gas - solido QS dT QS dP = ln P = - + cost dT R T2 RT QS = Calore latente di sublimazione Equilibrio liquido- solido dP QF = QF, calore latente di fusione, > 0 dT (VL - VS) T
 T.")
93
Caso 1° Caso 2° P P L L S S V V T T dP dT VL > VS , allora > 0,
La temperatura di fusione CRESCE con la pressione dP dP Caso 2° VL < VS , allora < 0, < 0 dT dT La temperatura di fusione DECRESCE con la pressione Caso 1° Quasi tutte le sostanze Caso 2° Poche sostanze (H2O) P S L V P T L S V T
 P. S. L. V. P. T. L. S. V. T.")
94
Principio di Le Chatelier o dell’equilibrio mobile
Quando si applica una modifica/spinta esterna ad un sistema in equilibrio, esso reagirà in modo da opporsi alla variazione apportata, per minimizzarne gli effetti (Henri Le Châtelier, 1884). Il principio di Le Chatelier consente di prevedere in modo estremamente semplice la direzione in cui evolve un sistema inizialmente all'equilibrio in seguito ad una data perturbazione.
. Il principio di Le Chatelier consente di prevedere in modo estremamente. semplice la direzione in cui evolve un sistema inizialmente all equilibrio. in seguito ad una data perturbazione.")
95
Equilibrio fisico liquido-vapore
diminuendo il volume del recipiente a T costante si osserverà uno spostamento del sistema verso lo stato liquido (condensazione del vapore) che è lo stato che occupa il minor volume. In tal modo il sistema all’equilibrio reagisce in modo da annullare, per quanto possibile, la perturbazione esterna (aumento della P).
 che è lo stato che occupa il minor volume. In tal modo il sistema all’equilibrio reagisce in modo da annullare, per quanto possibile, la perturbazione esterna (aumento della P).")
96
Un altro modo per perturbare un sistema all’equilibrio consiste
nel variare la sua T. Ricordiamo che : - reazioni endotermiche avvengono con acquisto di calore dall’ambiente (ΔH>0); - reazioni esotermiche avvengono con perdita di calore, cioè cedono calore all’ambiente (ΔH<0). L’aumento di T di una miscela all’equilibrio mediante riscaldamento fa sì che la reazione si sviluppi in modo tale da consumare parte del calore addizionato PERTANTO: - in una reazione endotermica l’equilibrio si sposta da sinistra verso destra (favoriti i prodotti: Keq aumenta con aumento di T) ; - in una reazione esotermica da destra a sinistra (favoriti i reagenti: Keq diminuisce con aumento di T).
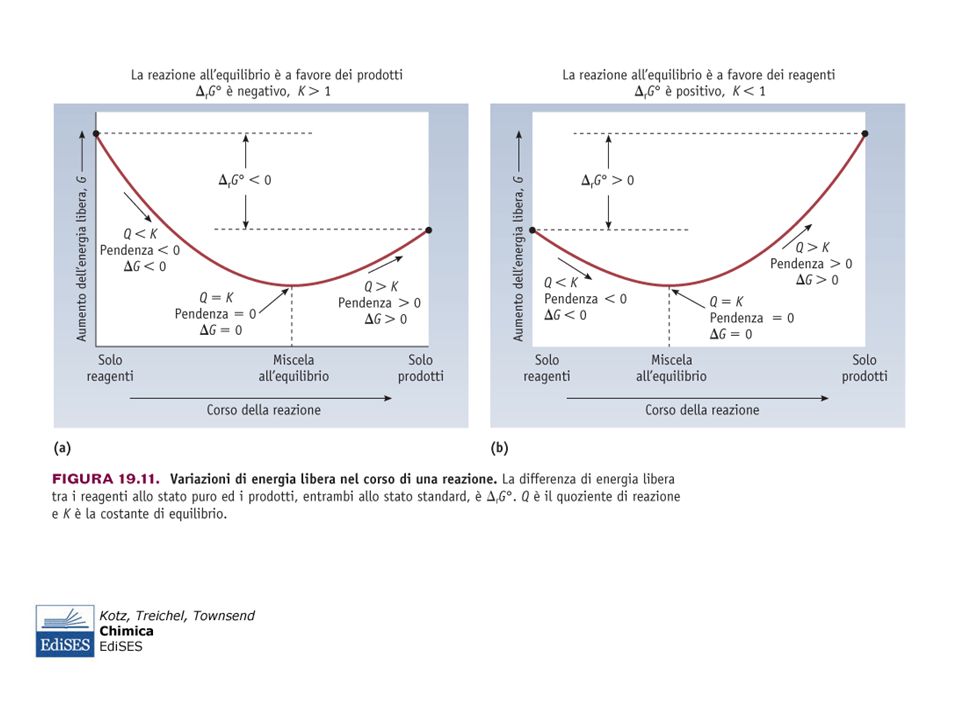
; - reazioni esotermiche avvengono. con perdita di calore, cioè cedono calore all’ambiente (ΔH<0). L’aumento di T di una miscela all’equilibrio mediante riscaldamento. fa sì che la reazione si sviluppi in modo tale da consumare parte del calore addizionato. PERTANTO: - in una reazione endotermica l’equilibrio si sposta da sinistra. verso destra (favoriti i prodotti: Keq aumenta con aumento di T) ; - in una reazione esotermica da destra a sinistra. (favoriti i reagenti: Keq diminuisce con aumento di T).")
97
La maggior parte delle reazioni di dissoluzione di composti ionici
è endotermica (ΔH>0) : per il principio di Le Chatelier, le solubilità in tali casi crescono con l’aumento della T. Nelle dissoluzioni esotermiche (ad es. caso di CaSO4) si osserva un comportamento opposto: le solubilità decrescono con l’aumento della T.
 : per il principio di Le Chatelier, le solubilità in tali casi crescono con l’aumento della T. Nelle dissoluzioni esotermiche (ad es. caso di CaSO4) si osserva un comportamento opposto: le solubilità decrescono con l’aumento della T.")
98
pres def VI -5.ppt

99
Proprietà colligative delle soluzioni
760 Dtcr = abbassamento del punto di fusione H2O (l) Sostanza Kcr Acqua = 1,86°C/mole Benzene = 5,12° C/mole Canfora = 40° C/mole H2O (s) soluzione Dteb = innalzamento del punto di ebollizzione temperatura Dtcr = Kcr m Dteb = Keb m Dtcr Dteb p = pressione osmotica VUOTO membrana Soluz. diluita H2O H2O Soluz. diluita EVAPORAZIONE OSMOSI
 Sostanza Kcr. Acqua = 1,86°C/mole. Benzene = 5,12° C/mole. Canfora = 40° C/mole. H2O (s) soluzione. Dteb = innalzamento del punto di. ebollizzione. temperatura. Dtcr = Kcr m. Dteb = Keb m. Dtcr. Dteb. p = pressione. osmotica. VUOTO. membrana. Soluz. diluita. H2O. H2O. Soluz. diluita. EVAPORAZIONE. OSMOSI.")
100
pres def VI - 6.ppt

101
SISTEMI A DUE COMPONENTI
Variabili: P, T, composizione T (P) P (T) Riportando sulle ordinate T (a P = cost) si ha un diagramma ISOBARO Riportando sulle ordinate P (a T = cost) si ha un diagramma ISOTERMO 0,75 moli B 0,25 moli A A B 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,0 XB 1,0 0,9 0,8 0,7 0,6 0,5 0,4 0,3 0,2 0,1 XA P P P°A T = Cost. LEGGI DI RAOULT: PA = P°A XA PB = P°B XB Ptot = PA + PB Ptot P°B PA PB A B
 P (T) Riportando sulle. ordinate T. (a P = cost) si ha. un diagramma. ISOBARO. Riportando sulle. ordinate P. (a T = cost) si ha. un diagramma. ISOTERMO. 0,75 moli B. 0,25 moli A. A. B. 0,1. 0,2. 0,3. 0,4. 0,5. 0,6. 0,7. 0,8. 0,9. 1,0. XB. 1,0. 0,9. 0,8. 0,7. 0,6. 0,5. 0,4. 0,3. 0,2. 0,1. XA. P. P. P°A. T = Cost. LEGGI DI RAOULT: PA = P°A XA. PB = P°B XB. Ptot = PA + PB. Ptot. P°B. PA. PB. A. B.")
102
pres def VI - 7.ppt

103
T (P cost) TA VAPORE LIQUIDO VAPORE TB LIQUIDO A XB B
(B) VAPORE DISTILLAZIONE FRAZIONATA T (P cost) TA VAPORE LIQUIDO VAPORE TB LIQUIDO A XB B (A) LIQUIDO
 VAPORE. DISTILLAZIONE FRAZIONATA. T. (P cost) TA. VAPORE. LIQUIDO. VAPORE. TB. LIQUIDO. A. XB. B. (A) LIQUIDO.")
104
Miscele Azeotropiche A B H2O C6H6O
- Quando A e B sono in soluzione alla composizione azeotropica Xc, essi non possono essere separati uno dall’altro mediante distillazione. - La miscela azeotropica bolle come un composto puro ad una T cost senza intervallo di ebollizione. TA TA V V L +V TB L +V L +V 78,5°C L C L A B H2O C6H6O XC X XB C2H6O

105
Equilibri tra due fasi liquide
Non sempre due liquidi sono totalmente miscibili, ma solo parzialmente X1 B A Tc XB b a (P = cost) T1 X2 (P = cost) 1 2 3 T'c T1 2 fasi liquide 2 fasi liquide T''c A B X1 X2 lacuna di solubilità XB Tc = temperatura critica di solubilità A T >Tc il sistema è omogeneo a qualunque composizione A T1<Tc il sistema è omogeneo solo se la sua composizione è esterna alla curva
 T1. X2. (P = cost) T c. T1. 2 fasi liquide. 2 fasi liquide. T c. A. B. X1. X2. lacuna di solubilità. XB. Tc = temperatura critica di solubilità. A T >Tc il sistema è omogeneo a qualunque composizione. A T1<Tc il sistema è omogeneo solo se la sua composizione è esterna alla curva.")
106
pres def VI - 8.ppt

107
Equilibri di Partizione
Soluto che si ripartisce tra due liquidi tra loro non miscibili. Si distribuisce in entrambi a seconda della solubilità in ciascuno di essi Liquido 1 C1 = K (Costante di partizione) Liquido 2 C2 Componenti = 3 Fasi = 2 V = C F = = 3 Se si fissano P, T, C1, deve essere fissa C2 Il valore di K è costante a P, t costanti
 Liquido 2. C2. Componenti = 3. Fasi = 2. V = C F = = 3. Se si fissano P, T, C1, deve essere fissa C2. Il valore di K è costante a P, t costanti.")
108
Solubilità dei Gas (Legge di Henry)
Applicando al gas disciolto la legge di Raoult PB = P°BXB; XB = 1 P°B PB P Gas "La frazione molare del gas nella soluzione è proporzionale alla pressione parziale del gas in equilibrio con la soluzione" Soluzione T = Cost In soluzione diluita: gB nB nB MB ~ XB = = = Cost. gB nA + nB nA nA XB gB = = Cost.' • PB Cost. Il peso del gas che si scioglie in un dato volume di solvente è proporzionale alla pressione del gas.

Presentazioni simili















 Acidi poliprotici>")










>")

