Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

6
Sindrome di Myhre Bassa statura Rigidità articolare Ipertrofia muscolare generalizzata Serie riconoscibile di caratteristiche facciali: (fessure palpebrali corte, ipoplasia della parte mediana della faccia, bocca piccola con un sottile labbro superiore, prognatismo) Anomalie scheletriche: (ossa del cranio spesse, platispondilia, coste allargate, ali iliache ipoplastiche e brachidattilia) Deficit cognitivi variabili Numero di casi annotati <30 Eredità autosomica dominante per insorgenza de novo? Caratteristiche cliniche di pazienti rappresentativi affetti da sindrome di Myhre

7
Scopo del lavoro 2. Valutazione dell’origine parentale della mutazione 1. Identificazione dell’evento molecolare responsabile della sindrome Obiettivi della tesi.

8
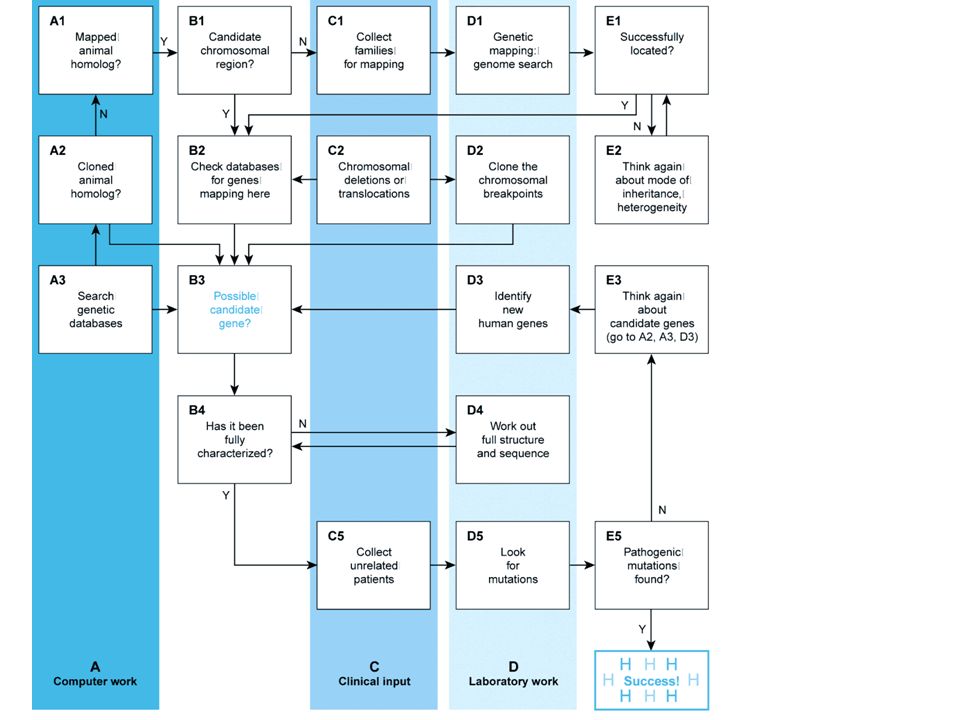
1 Estrazione del DNA 2 Sequenziamento dell’esoma di un singolo paziente 3 Filtraggio delle varianti 4 Prioritizzazione delle varianti 5 Validazione delle varianti 6 C o n f e r m a d e ll a m u t a z i o n e n e l p r o b a n d o e i n a l t r i 7 a f f e t t i 7 Ricerca di famiglie informative 8 Analisi degli aplotipi Passi sperimentali Diagramma di flusso della procedura sperimentale utilizzata.

9
Composizione del campione di studio 8 soggetti con caratteristiche cliniche coerenti con la MS e i loro genitori La raccolta e l’archiviazione del materiale biologico sono state possibili grazie alla collaborazione con i centri di provenienza dei campioni: - Ospedale “Bambino Gesù” (Roma), Prof. Dallapiccola - Università di Torino, Prof.ssa Silengo - Arcispedale Santa Maria Nuova (Reggio Emilia), Dott.ssa Garavelli - Ospedale Microcitemico (Cagliari), Dott.ssa Boccone - Università “Federico II” (Napoli), Dott.ssa Melis
, Dott.ssa Garavelli - Ospedale Microcitemico (Cagliari), Dott.ssa Boccone - Università Federico II (Napoli), Dott.ssa Melis.")
10
Sequenziamento dell’esoma 1. Preparazione della libreria di sstDNA 2. Cattura dell’esoma 3. Processamento della libreria 4. Sequenziamento della libreria 5. Analisi dei dati ottenuti Due sequenziamenti separati di un singolo paziente affetto Genome sequencer 454 GS FLX Titanium (Roche), Istituto Superiore di Sanità, Reparto di fisiopatologia delle malattie genetiche Cattura dell’esoma mediante Sequence Capture Human Exome 2.1M Array v.2 (Nimblegen) Flusso di lavoro del sequenziatore 454 (Roche).
, Istituto Superiore di Sanità, Reparto di fisiopatologia delle malattie genetiche Cattura dell’esoma mediante Sequence Capture Human Exome 2.1M Array v.2 (Nimblegen) Flusso di lavoro del sequenziatore 454 (Roche)..")
11
Risultati del sequenziamento Lunghezza media delle read: 380 bp Copertura delle sequenze bersaglio: 98% Profondità media di sequenza: 10x Fluogramma prodotto dal software del sequenziatore. I fluogrammi provenienti da ogni pozzetto della piastra PTP vengono interpretati, ottenendo così le reads che verranno mappate sul genoma di riferimento al fine di individuare le zone di identità e le posizioni varianti. Assemblaggio Mappatura Analisi delle varianti

12
Filtraggio delle varianti AllDiffs BEDTools 133.889 GSMapper HCDiffs dbSNP 72.426 varianti 665 nuove varianti Uniprot, HapMap codificanti BLAST 185 varianti non sinonime 18 varianti di splicing Progressiva riduzione delle varianti prodotte dal sequenziamento dell’esoma ottenuta attraverso l’applicazione sequenziale dei diversi tools informatici.

13
Prioritizzazione delle varianti filtrate Due approcci SIFT PolyPhen-2 Varianti causali candidate ADAMTS3 PAPLN HKR1 DUSP16 BRD7 ARHGAP5 SNX10 HAS3 GLI1 PP3CA Metodo “soggettivo”Metodo “oggettivo” 203 varianti Rappresentazione schematica della scelta delle varianti da validare effettuata attraverso approccio soggettivo ed oggettivo. TGF- β DAVID Génie Varianti causali candidate DAVID Génie PITX2 CRB1 SMAD4 EFHC1 THBS3 HSPG2 MYH2 PKD2 PITX2 SETX MRE11A SMAD4 USH1C Sostituzion e deleteria

14
Validazione delle varianti selezionate Le regioni codificanti colpite dalle mutazioni selezionate sono state confermate nel paziente e poi verificate nei genitori tramite sequenziamento con metodo di Sanger. Validazioni metodo “soggettivo” Validazioni metodo “oggettivo”

15
Coinvolgimento causale del gene SMAD4 Risultati L’esone 11 del gene è stato sequenziato anche a partire dai campioni di DNA estratti da altri tessuti escludendo fortemente la possibilità di un evento somatico. L’intera sequenza codificante del gene è stata sequenziata anche negli altri sette individui affetti non imparentati. Identificazione di due mutazioni de novo in eterozigosi che colpiscono il medesimo codone: c.1498A>G e c.1499T>C Elettroferogrammi che mostrano la presenza delle sostituzioni non sinonime in eterozigosi in vari tessuti di due pazienti.

16
Varianti identificate nel gene SMAD4 Risultati 2 mutazioni de novo identificate nell’intera coorte (8 casi) Sostituzione del medesimo amminoacido isoleucina500 con una valina (5 casi) o con una treonina (3 casi)
 Sostituzione del medesimo amminoacido isoleucina500 con una valina (5 casi) o con una treonina (3 casi)")
17
SMAD4 Discussione E’ un membro della famiglia di fattori di trascrizione intracellulari che operano nella via di segnalazione del TGF- β https://en.wikipedia.org/wiki/Transforming_growth_factor_beta 3 classi: R-Smad, I-Smad e Co-Smad (SMAD4) Due domini: MH1 e MH2 uniti da una regione linker Le 3 classi della famiglia di proteine Smad e i loro elementi strutturali (Massagué et al., 2005).
 Due domini: MH1 e MH2 uniti da una regione linker Le 3 classi della famiglia di proteine Smad e i loro elementi strutturali (Massagué et al., 2005).")
18
Via di segnalazione del TGF-β I ligandi della superfamiglia del TGF- β iniziano la segnalazione assemblando dei complessi recettoriali che attivano i fattori trascrizionali SMAD. SMAD4 è essenziale per la formazione dei complessi trascrizionalmente attivi. La sua disregolazione è attesa avere effetti pleiotropici. Trasduzione del segnale nel pathway del TGF-β

19
Conservazione del residuo Ile500 del dominio MH2 Il residuo Ile500 mutato nella MS ricade all’interno del dominio MH2, il quale risulta altamente conservato tra gli ortologhi e paraloghi. Allineamento parziale della sequenza amminoacidica del dominio MH2 di SMAD4 (dal residuo 489 al 552) negli ortologhi. Si può notare la conservazione sia del residuo Ile500 (freccia) che dei residui colpiti da mutazioni somatiche associate al cancro (cerchi rossi) e/o dalle lesioni germinali che ricorrono nella JPS e nella JPS-HHT (cerchi blu).
 negli ortologhi. Si può notare la conservazione sia del residuo Ile500 (freccia) che dei residui colpiti da mutazioni somatiche associate al cancro (cerchi rossi) e/o dalle lesioni germinali che ricorrono nella JPS e nella JPS-HHT (cerchi blu)..")
20
SMAD4 oncosoppressore Mutazioni somatiche: colpito da perdita biallelica in tumori pancreatici, tumori metastatici del colon e in altri carcinomi Mutazioni germinali: nella JPS e nella JPS-HHT Nessun coinvolgimento della Ile500 Rappresentazione schematica del gene SMAD4. Sono mostrate le mutazioni non senso (cerchi rossi) e le delezioni (triangoli rosa) riportate nella JPS e nella JPS-HHT. I picchi blu indicano la distribuzione delle mutazioni somatiche ricorrenti nel cancro.
 e le delezioni (triangoli rosa) riportate nella JPS e nella JPS-HHT. I picchi blu indicano la distribuzione delle mutazioni somatiche ricorrenti nel cancro..")
21
Possibili effetti promossi dalle mutazioni della MS Compromettono il legame di SMAD4 ai suoi partner di segnalazione Impediscono la corretta ubiquitinazione sulla Lys519 e perturbano il flusso del segnale (A) Rappresentazione della superficie accessibile al solvente del complesso formato dai domini MH2 di SMAD4/SMAD3. Il monomero SMAD4 è mostrato in azzurro, mentre le due subunità SMAD3 sono colorate in giallo e bianco. Il residuo Ile500 è colorato in rosso. (B) Rappresentazione dell’interazione tra la Ile500 (rosso) e i residui circostanti. In blu: Arg496, Arg497, Arg502, Glu526, e His528. In viola: Lys519.
 Rappresentazione dell’interazione tra la Ile500 (rosso) e i residui circostanti. In blu: Arg496, Arg497, Arg502, Glu526, e His528. In viola: Lys519..")
22
Origine parentale delle mutazioni puntiformi Divisioni delle cellule germinali nella linea maschile. S cellula staminale; G cellula gonadica; M cellula meiotica (Strachan and Read, 2006) Il numero di divisioni cellulari necessarie per produrre gli spermatozoi è in relazione con l’età. Errori casuali di copiatura del DNA tendono ad insorgere principalmente nella linea germinale maschile. In più: ridotta fedeltà nella replicazione del DNA, inefficienza dei meccanismi di riparazione, esposizione a mutageni, possibili eventi di selezione.
 Il numero di divisioni cellulari necessarie per produrre gli spermatozoi è in relazione con l’età. Errori casuali di copiatura del DNA tendono ad insorgere principalmente nella linea germinale maschile. In più: ridotta fedeltà nella replicazione del DNA, inefficienza dei meccanismi di riparazione, esposizione a mutageni, possibili eventi di selezione..")
23
Ricerca di famiglie informative 10 polimorfismi intronici 5 famiglie informative Clonaggio dei pazienti Analisi degli aplotipi \ Esempio di famiglia informativa e aplotipi attesi tramite clonaggio di un frammento di DNA genomico del probando contenente il polimorfismo intronico informativo e la mutazione.

24
Origine paterna della mutazione Aplotipi del probando determinati attraverso amplificazione e clonaggio della sequenza genomica comprendente il sito polimorfico e la mutazione. Aplotipo materno (WT) Aplotipo paterno (mutato)
 Aplotipo paterno (mutato).")
25
Conclusioni Un ristretto spettro di mutazioni de novo, non sinonime ed eterozigoti di SMAD4 sono state identificate in tutti i soggetti affetti da MS inclusi nello studio: disordine geneticamente omogeneo fenotipo relativamente uniforme Eredità paterna in 5 casi su 5 sbilanciamento a favore dell’origine paterna osservato in molte malattie dominanti dovute a mutazioni puntiformi. SMAD4 è il gene responsabile della sindrome di Myhre Caratteristiche cliniche di pazienti con mutazioni eterozigoti in SMAD4 (Le Goff et al., 2012)
.")
Presentazioni simili








, L Garagnani (2), L Schirosi (3), C De Gaetani (4), A Maiorana.>")








>")


