Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Introduzione all’analisi cromatografica

2
La cromatografia è un metodo analitico largamente utilizzato per la separazione, l’identificazione e la determinazione quantitativa dei componenti di miscele complesse I metodi cromatografici sfruttano le diverse affinità delle molecole nei confronti di due fasi diverse: la fase stazionaria e la fase mobile che viene fatta scorrere in modo continuo sulla fase stazionaria I componenti della miscela sono trasportati attraverso la fase stazionaria dalla fase mobile La separazione dei componenti della miscela dipende dalla loro differente velocità di migrazione. In particolare le sostanze più affini alla fase stazionaria sono più trattenute e quindi il loro trascinamento risulta lento. Al contrario sostanze più affini alla fase mobile sono più facilmente eluite

3
Fase mobile Fase stazionaria

4
Da un punto di vista storico, i primi esperimenti cromatografici furono eseguiti dal botanico russo M Tswett nel 1906 (separazione di clorofille usando etere di petrolio come fase eluente e calcio carbonato come fase stazionaria). Le specie separate apparvero come bande colorate e da qui il nome che scelse per la tecnica: dal Greco chroma (colore) e graphein (scrivere)
 e graphein (scrivere)")
5
Cromatografia liquida
Classificazione dei metodi cromatografici Un metodo di classificazione dei metodi cromatografici si basa sullo stato della fase mobile (liquido – gassoso) cromatografia Cromatografia liquida (fase mobile liquida) Gas cromatografia (fase mobile gassosa) Sulla base dello stato della fase stazionaria si ha una ulteriore suddivisione liquido – liquido liquido - solida gas – liquido gas - solida
 cromatografia. Cromatografia liquida. (fase mobile liquida) Gas cromatografia. (fase mobile gassosa) Sulla base dello stato della fase stazionaria si ha una ulteriore suddivisione. liquido – liquido. liquido - solida. gas – liquido. gas - solida.")
6
Fase stazionaria (solido)
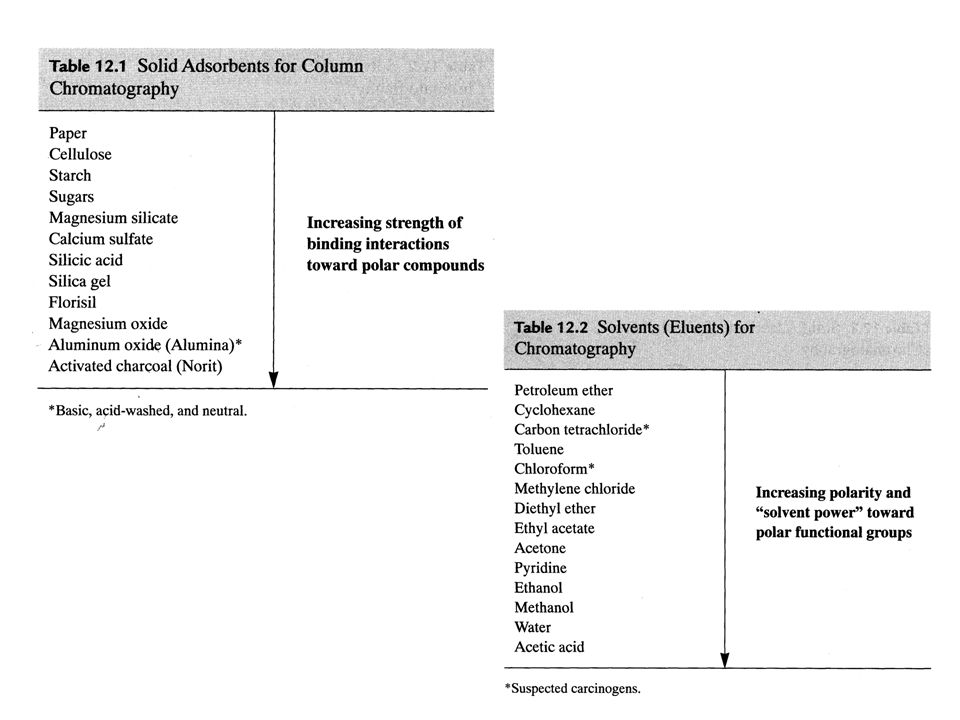
Le tecniche cromatografiche vengono inoltre classificate in base al meccanismo principale di separazione che interviene: 1) adsorbimento, 2) ripartizione, 3) scambio ionico, 4) esclusione, 5) affinità 1. Adsorbimento: la fase stazionaria è un solido in granuli e la fase mobile può essere un gas o un liquido. La fase stazionaria è in genere un composto inorganico o un polimero organico. Sulla superficie dei granuli si trovano dei siti attivi che possono stabilire legami deboli (interazioni dipolo-dipolo, legami idrogeno, interazione idrofobiche) con le molecole della miscela Fase mobile Fase stazionaria (solido)
 adsorbimento, 2) ripartizione, 3) scambio ionico, 4) esclusione, 5) affinità. 1. Adsorbimento: la fase stazionaria è un solido in granuli e la fase mobile può essere un gas o un liquido. La fase stazionaria è in genere un composto inorganico o un polimero organico. Sulla superficie dei granuli si trovano dei siti attivi che possono stabilire legami deboli (interazioni dipolo-dipolo, legami idrogeno, interazione idrofobiche) con le molecole della miscela. Fase mobile. Fase stazionaria (solido)")
7
Possibili interazioni di composti organici con allumina

9
Fase stazionaria (liquido)
2. Ripartizione: la fase stazionaria è un liquido (supportato da un solido granulare inerte) in cui le sostanze da separare si solubilizzano. La fase mobile può essere un gas o un liquido e comunque deve essere immiscibile con la fase liquida. Durante l’eluizione le molecole si ripartiscono dinamicamente nelle due fasi (stazionaria e mobile) sulla base delle caratteristiche chimiche Fase mobile Fase stazionaria (liquido) supporto
 in cui le sostanze da separare si solubilizzano. La fase mobile può essere un gas o un liquido e comunque deve essere immiscibile con la fase liquida. Durante l’eluizione le molecole si ripartiscono dinamicamente nelle due fasi (stazionaria e mobile) sulla base delle caratteristiche chimiche. Fase mobile. Fase stazionaria (liquido) supporto.")
10
Fase mobile - controioni - - - - - - + + + Siti attivi + + +
3. Scambio ionico:la fase stazionaria è costituita da macromolecole con siti attivi ionizzati (cationici o anionici) i cui controioni possono essere scambiati con ioni aventi carica dello stesso segno ed eluiti dalla fase mobile. Durante l’eluizione gli ioni presenti nella fase mobile vengono separati gli uni dagli altri in base alla diversa affinità di ciascuno per i siti della fase stazionaria (cromatografia a scambio ionico) Fase mobile - controioni - - - - - - + + + Siti attivi + + + Fase stazionaria Fase stazionaria - - - - - - - - + + + + + + Fase stazionaria Fase stazionaria
 i cui controioni possono essere scambiati con ioni aventi carica dello stesso segno ed eluiti dalla fase mobile. Durante l’eluizione gli ioni presenti nella fase mobile vengono separati gli uni dagli altri in base alla diversa affinità di ciascuno per i siti della fase stazionaria (cromatografia a scambio ionico) Fase mobile. - controioni Siti attivi Fase stazionaria. Fase stazionaria Fase stazionaria. Fase stazionaria.")
11
4. Esclusione: tecnica per la separazione di macromolecole
4. Esclusione: tecnica per la separazione di macromolecole. La fase stazionaria è un solido poroso o, più comunemente, un gel con i pori di dimensioni variabili. Le molecole dell’analita di una determinata dimensione molecolare, penetrano nei pori dove permangono per un certo periodo; le molecole troppo grandi, però, vengono escluse dai pori e perciò escono dalla colonna in tempi brevi (cromatografia di esclusione)
")
12
Tecniche cromatografiche
Cromatografia planare: la fase stazionaria è distribuita su una superficie piana. Le tecniche principali sono la cromatografia su strato sottile (Thin Layer Chormagraphy, TLC) e la cromatografia su carta (paper chromatography)
 e la cromatografia su carta (paper chromatography)")
13
Cromatografia su colonna a bassa pressione : ampiamente utilizzata per separazione di miscele e non per analisi quantitativa

14
Cromatografia in fase liquida ad elevata prestazione (High Performance Liquid Chromatography, HPLC): ampiamente utilizzata per l’identificazione e quantizzazione di molecole. In pratica, la versione strumentale della cromatografia su colonna 1) Fase eluente; 3) sistema di pompaggio; 4) sistema iniezione campione; 5) colonna cromatografica; 6) detector ; 7) scarico; 8) sistema acquisizione dati
 Fase eluente; 3) sistema di pompaggio; 4) sistema iniezione campione; 5) colonna cromatografica; 6) detector ; 7) scarico; 8) sistema acquisizione dati.")
15
Colonne HPLC

16
Gascromatografia (Gas Chromatography, GC): Tecnica ampiamente utilizzata per l’identificazione e quantizzazione di molecole volatili.
: Tecnica ampiamente utilizzata per l’identificazione e quantizzazione di molecole volatili.")
17
Colonna GC (capillare)
")
18
Il cromatogramma Le separazioni cromatografiche strumentali (HPLC e GC) si concludono con la registrazione del cromatogramma, ovvero del tracciato del segnale del rivelatore in funzione del tempo o del volume di eluente, a partire dall’istante in cui la miscela viene introdotta nella colonna (t=0)
 si concludono con la registrazione del cromatogramma, ovvero del tracciato del segnale del rivelatore in funzione del tempo o del volume di eluente, a partire dall’istante in cui la miscela viene introdotta nella colonna (t=0)")
19
Il tracciato completo che si ottiene per ciascuna sostanza eluita è detto picco cromatografico. In condizioni cromatografiche ideali il picco cromatografico ha la forma di una curva gaussiana. Da un punto di vista matematico una curva gaussiana viene descritta dal punto di massimo e dalla distanza tra i punti di flesso (tale distanza diviso 2 è la deviazione std. () Altezza del picco: distanza tra il punto massimo e la tangente alla linea di base Larghezza della base del picco (W): lunghezza del segmento interpolato all’intersezione fra le tangenti ai flessi della gaussiana e la linea di base (Wb = 4 ) Larghezza a metà altezza: larghezza misurata a metà altezza del picco (WH = 2.354) Distanza fra i punti di flesso: segmento interpolato fra i due punti di flesso (corrsipondente al 60.7% altezza picco) (Wi = 2 ) Area totale sottesa: proporzionale alla concentrazione
: lunghezza del segmento interpolato all’intersezione fra le tangenti ai flessi della gaussiana e la linea di base (Wb = 4 ) Larghezza a metà altezza: larghezza misurata a metà altezza del picco (WH = 2.354) Distanza fra i punti di flesso: segmento interpolato fra i due punti di flesso (corrsipondente al 60.7% altezza picco) (Wi = 2 ) Area totale sottesa: proporzionale alla concentrazione.")
20
Il picco cromatografico è inoltre identificato dal tempo di ritenzione (tR): tempo impiegato da ciascuna sostanza per eluire dalla colonna, misurato a partire dall’istante in cui la miscela viene introdotta nello strumento, fino all’istante in cui si registra il massimo del picco (in alternativa si può utilizzare il volume di ritenzione). Il tempo morto è il tempo di ritenzione di una sostanza non trattenuta dalla fase stazionaria Il volume morto (VM) (detto anche volume della fase mobile) corrisponde al volume della colonna non occupato dalla fase stazionaria. Il tempo di ritenzione corretto: tempo che ogni sostanza impiega per le interazioni con la fase stazionaria (t’R = tR – tM)
 (detto anche volume della fase mobile) corrisponde al volume della colonna non occupato dalla fase stazionaria. Il tempo di ritenzione corretto: tempo che ogni sostanza impiega per le interazioni con la fase stazionaria (t’R = tR – tM)")
21
CM CS VR = VM + KCVS Costante di distribuzione CS
Ad ogni istante del processo cromatografico le sostanze si distribuiscono fra le due fasi in modo specifico. Supponiamo che ad un tempo t si raggiunga l’equilibrio: CM CS ove Cs= concentrazione analita nella fase stazionaria e CM = conc nella fase mobile; CS CM Costante di distribuzione (Kc) = La costante di distribuzione è correlata con il volume di ritenzione dalla seguente equazione: VR = VM + KCVS Ove VS è il volume della fase stazionaria. Il valore KC può quindi essere determinato sulla base di VR, VM e VS. Dato tuttavia la difficoltà di determinare in modo accurato VS, si preferisce far riferimento al fattore di ritenzione (k)
 = La costante di distribuzione è correlata con il volume di ritenzione dalla seguente equazione: VR = VM + KCVS. Ove VS è il volume della fase stazionaria. Il valore KC può quindi essere determinato sulla base di VR, VM e VS. Dato tuttavia la difficoltà di determinare in modo accurato VS, si preferisce far riferimento al fattore di ritenzione (k)")
22
Fattore di ritenzione o fattore di capacità (k) =
nS nM Fattore di ritenzione o fattore di capacità (k) = Ove nS è il numero di moli nella fase stazionaria e nM nella fase mobile CSVS VS VM Poiché KC= CS/CM k = k = KC KC = k CMVM VM VS Sostituendo KC nella equazione VR = VM + KCVS avremo: VR = VM + kVM
 = Ove nS è il numero di moli nella fase stazionaria e nM nella fase mobile. CSVS. VS. VM. Poiché KC= CS/CM. k = k = KC. KC = k. CMVM. VM. VS. Sostituendo KC nella equazione VR = VM + KCVS avremo: VR = VM + kVM.")
23
tRFC = tMFC (1+k) tR = tM (1+k) k= (tR-tM)/tM ovvero: t’R k = tM
e quindi VR = VM (1 + k) se esprimiamo tale equazione in termini di tempi di ritenzione (V= TF) si può scrivere: tRFC = tMFC (1+k) e considerando il flusso costante, L’equazione si semplifica tR = tM (1+k) da cui k= (tR-tM)/tM ovvero: t’R tM k =
 se esprimiamo tale equazione in termini di tempi di ritenzione (V= TF) si può scrivere: tRFC = tMFC (1+k) e considerando il flusso costante, L’equazione si semplifica. tR = tM (1+k) da cui. k= (tR-tM)/tM ovvero: t’R. tM. k =")
24
Al fine di una buona separazione è necessario che il fattore di ritenzione della sostanza eluita per prima sia maggiore di 1. k non deve inoltre essere superiore a 10-15

25
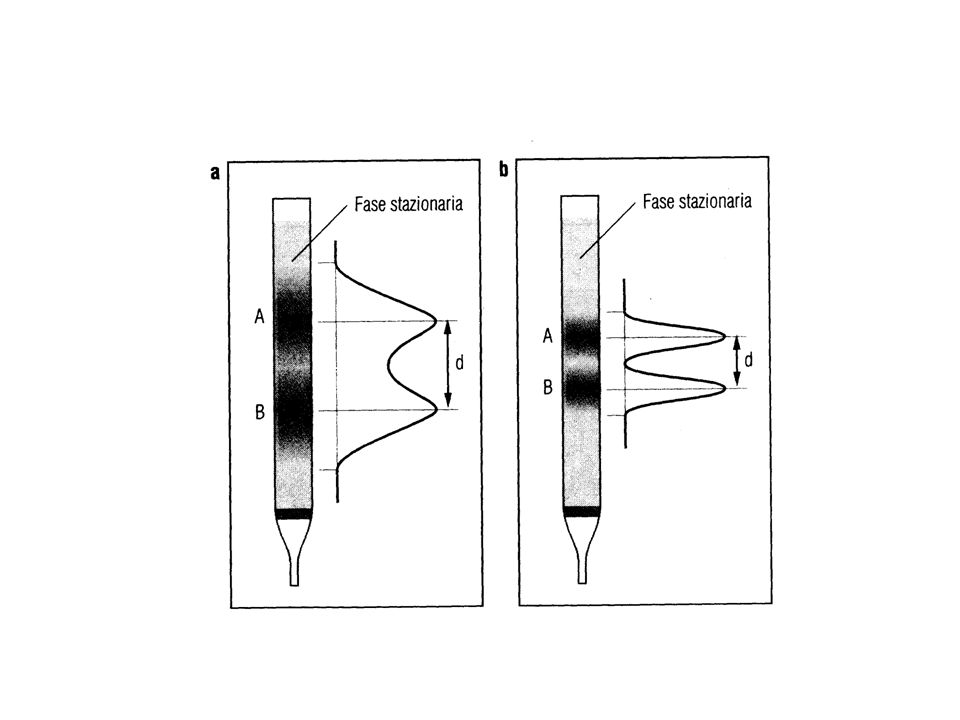
Risoluzione di una separazione cromatografica
Una buona risoluzione cromatografica significa una buona separazione tra i due picchi contigui selettività efficienza

26
fattore di separazione = t’R2 / t’R1
La qualità di una separazione cromatografica (risoluzione dei picchi) dipende dalla selettività ed efficienza: la selettività indica la capacità di un sistema cromatografico di eluire specie chimiche a velocità il più possibili diverse ed è espressa dal fattore di separazione = t’R2 / t’R1 La selettività non è tuttavia suff. per una buona risoluzione cromatografica qualora i picchi sono larghi e tali da sovrapporsi
 dipende dalla selettività ed efficienza: la selettività indica la capacità di un sistema cromatografico di eluire specie chimiche a velocità il più possibili diverse ed è espressa dal. fattore di separazione = t’R2 / t’R1. La selettività non è tuttavia suff. per una buona risoluzione cromatografica qualora i picchi sono larghi e tali da sovrapporsi.")
27
Efficienza della colonna N = (tR/ )2 = 16 (tR/ wb)2
L’efficienza indica la capacità di un sistema cromatografico di eluire tutte le particelle di una specie chimica con la stessa velocità, in modo da formare bande e quindi picchi stretti. Dato che l’efficienza esprime l’allargamento del picco essa è una espressione di : Deviazione std. relativa R = /tR, dato che tale valore è solitamente piccolo, per motivi pratici, si preferisce calcolare N, detto numero dei piatti o numero dei piatti teorici: Efficienza della colonna N = (tR/ )2 = 16 (tR/ wb)2
2 = 16 (tR/ wb)2.")
29
Valutazione quantitativa dell’ efficienza di una colonna cromatografica
Altezza equivalente al piatto teorico H = L / N Ove L è la lunghezza della colonna (una colonna è tanto più efficiente quanto più H è piccolo) L’efficienza di una colonna dipende da fattori extra-colonna (non incidono più del 10%) e da fattori della colonna. Dei diversi modelli utilizzati per spiegare i fattori della colonna, quello più accreditato è il modello del non-equilibrio di Giddings che individua tre fattori che partecipano all’allargamento delle bande: Percorsi multipli diffusione molecolare longitudinale distribuzione di flusso Trasferimento di massa tra le fasi
 L’efficienza di una colonna dipende da fattori extra-colonna (non incidono più del 10%) e da fattori della colonna. Dei diversi modelli utilizzati per spiegare i fattori della colonna, quello più accreditato è il modello del non-equilibrio di Giddings che individua tre fattori che partecipano all’allargamento delle bande: Percorsi multipli. diffusione molecolare longitudinale. distribuzione di flusso. Trasferimento di massa tra le fasi.")
30
Percorsi multipli: dato che la porosità e l’impaccamento della fase stazionaria non sono perfettamente uniformi, si verificano cammini diversi e non equivalenti dell’analita nella fase mobile. Questo comporta che le molecole, pur partendo insieme, percorrano distanze diverse e arrivano al detector a tempi diversi, provocando un allargamento della banda. Per risolvere tale problema è necessario ottenere una distribuzione granulometrica della fase stazionaria molto ristretta.

31
Diffusione di flusso: Il flusso è più veloce al centro che vicino alle particelle
L’allargamento da percorsi multipli e da diffusione di flusso è indipendente dalla velocità della fase mobile.

32
Diffusione molecolare longitudinale: Le molecole della sostanza si muovono spontaneamente in direzione longitudinale, sia nel senso di scorrimento della fase mobile sia in quello opposto. L’allargamento della banda per effetto della diffusione molecolare dipende dal tempo di permanenza dell’analita nella fase mobile e dalla diffusitività del soluto. Per minimizzare la diffusione molecolare longitudinale si può aumentare il flusso della fase mobile e agire sulla viscosità dell’eleuente

33
Trasferimento di massa: All’inizio le molecole di analita più vicine alla fase stazionaria diffondono dalla fase mobile alla fase stazionaria. La velocità di diffusione dipende dalla diffusività dell’analita. Le molecole rimaste nella fase mobile si spostano trascinate dalla corrente dell’eluente e diffondono nella fase stazionaria in un tratto successivo. Tale processo crea un allargamento della banda. Lo stesso meccanismo di diffusione asimmetrica si instaura per il procedimento inverso (dalla fase stazionaria alla fase mobile) Fase mobile Fase mobile Fase mobile Fase stazionaria Fase stazionaria Fase stazionaria Il trasferimento di massa è un processo che si riduce usando solventi a bassa viscosità (aumento della diffusività dell’analita) ed aumenta all’aumentare della velocità della fase mobile.
 ed aumenta all’aumentare della velocità della fase mobile.")
34
Variabili che influenzano l’efficienza della colonna: un modello matematico
L’ entità di allargamento di una banda dipende dal tempo in cui fase mobile e fase stazionaria restano in contatto, quindi dipende dalla velocità del flusso. Di conseguenza tutti gli studi di efficienza determinano H in funzione della velocità della fase mobile. Andamenti sperimentali tipici della funzione H= f(ū) ove ū = velocità lineare fase mobile (cm/s) Valore minimo di H (max di efficienza) per valori di ū bassi. H è di un unità di grandezza inferiore in a) ma le colonne non più lunghe di cm contro 50 m di b) quindi molto più efficienti in b).
 ove ū = velocità lineare fase mobile (cm/s) Valore minimo di H (max di efficienza) per valori di ū bassi. H è di un unità di grandezza. inferiore in a) ma le colonne non più lunghe di cm contro 50 m di b) quindi molto. più efficienti in b).")
35
L’equazione di Van Deemter
La funzione H= f(ū) può essere descritta da una equazione del tipo: y = a+ b/x + cx e può essere considerata la combinazione lineare di tre equazioni più semplici: y = a y= b/x (iperbole equilatera) y= cx y= cx y = a+ b/x + cx y= b/x y = a
 può essere descritta da una equazione del tipo: y = a+ b/x + cx. e può essere considerata la combinazione lineare di tre equazioni più semplici: y = a. y= b/x (iperbole equilatera) y= cx. y= cx. y = a+ b/x + cx. y= b/x. y = a.")
36
H = A + B/ū + C ū Equazione di Van Deemter descrive l’ efficienza
Cromatografica. H = A + B/ū + C ū Primo termine dell’equazione (A): A è associato ai percorsi multipli e alla diffusione di flusso (parametri indipendente dal flusso) A= 2dp Ove è un parametro associato alla granulometria (diametro e distribuzione) dp = diametro medio particelle Per minimizzare questo termine è necessario ridurre il diametro particelle e aumentarne l’uniformità
: A è associato ai percorsi multipli e alla diffusione di flusso (parametri indipendente dal flusso) A= 2dp. Ove è un parametro associato alla granulometria (diametro e distribuzione) dp = diametro medio particelle. Per minimizzare questo termine è necessario ridurre il diametro particelle e aumentarne l’uniformità.")
37
Secondo termine dell’equazione (B/ ū): Tale termine è associato alla diffusione molecolare longitudinale B= 2DM Ove è il fattore di tortuosità e dipende dall’impaccamento colonna DM è il coefficiente di diffusione del soluto nella fase mobile (in HPLC si può trascurare mentre è importante in GC) Per ridurre B si può ridurre (particelle uniformi), ridurre DM e aumentare ū
 Per ridurre B si può ridurre (particelle uniformi), ridurre DM e aumentare ū.")
38
qkdf2 CS= (1+k)2 DS dp2 CM= DM C = CS + CM
Terzo termine dell’equazione (C ū): Il terzo termine è associato alla resistenza al trasferimento di massa. C = CS + CM Ove CS è il contributo del trasferimento nella fase stazionaria e CM in quella mobile qkdf2 CS= (1+k)2 DS q = costante di uniformità particelle fase stazionaria, k= fattore di ritenzione; df = spessore massimo fase stazionaria; DS= coeff. di diffusione dp2 dp = spessore massimo fase mobile; DM= coeff. diffusione soluto nella fase mobile CM= DM
: Il terzo termine è associato alla resistenza al trasferimento di massa. C = CS + CM. Ove CS è il contributo del trasferimento nella fase stazionaria e CM in quella mobile. qkdf2. CS= (1+k)2 DS. q = costante di uniformità particelle fase stazionaria, k= fattore di ritenzione; df = spessore massimo fase stazionaria; DS= coeff. di diffusione. dp2. dp = spessore massimo fase mobile; DM= coeff. diffusione soluto nella fase mobile. CM= DM.")
39
Dall’equazione di Van Deemter si devono ottimizzare i seguenti parametri:
Flusso fase mobile : si effettua per via sperimentale e costituisce un “compromesso”; Caratteristiche fisiche fase mobile: importante per la GC (il termine B contribuisce notevolmente) – la fase gassosa deve avere una elevata densità diametro delle particelle della fase stazionaria – importante uniformità e ridotto diametro delle particelle
 – la fase gassosa deve avere una elevata densità. diametro delle particelle della fase stazionaria – importante uniformità e ridotto diametro delle particelle.")
40
tR2 – tR1 Rs= w1 + w2 2 Risoluzione
La risoluzione indica il grado di separazione dei picchi e dipende dalla selettività ed efficienza. In termini matematici la risoluzione tra due picchi adiacenti (Rs) è espressa dal rapporto fra le loro distanze e la semisomma delle rispettive larghezze alla base tR2 – tR1 w1 + w2 2 Rs=
 è espressa dal rapporto fra le loro distanze e la semisomma delle rispettive larghezze alla base. tR2 – tR1. w1 + w2. 2. Rs=")
41
Nel caso reale di picchi asimmetrici, l’equazione deve essere modificata ponendo al denominatore la somma dei segmenti a e b, ovvero le due semibasi che si affiancano tR2 – tR1 Rs= (a – b)
")
42
Il valore minimo di Rs è di 1 (sovrapposizione dei picchi del 4%)
Il valore minimo di Rs è di 1 (sovrapposizione dei picchi del 4%). Per Rs= 1.5 (sovrapposizione dello 0.3%) la separazione è considerata completa. La risoluzione può essere aumentata allungando la colonna quindi aumentando i piatti teorici.
. Per Rs= 1.5 (sovrapposizione dello 0.3%) la separazione è considerata completa. La risoluzione può essere aumentata allungando la colonna quindi aumentando i piatti teorici.")
43
La risoluzione fra due picchi può anche essere posta in relazione con il numero di piatti teorici (N), con la selettività () e con il fattore di ritenzione (k). Rs = 1 4 N2 - 1 k2 1+k2 ( ) Ove N2 e k2 si riferiscono al secondo picco (più lento) Variazioni N Variazione del fattore di ritenzione (k) : per fasi mobili gassose si varia la T, per quelle liquide la composizione dell’eluente Variazione del fattore selettività (): la variazione dei due parametri precedenti non è suff. nel caso in cui =1. In tale caso si variano diversi parametri tra cui la fase stazionaria, la fase mobile e la T
 Ove N2 e k2 si riferiscono al secondo picco (più lento) Variazioni N. Variazione del fattore di ritenzione (k) : per fasi mobili gassose si varia la T, per quelle liquide la composizione dell’eluente. Variazione del fattore selettività (): la variazione dei due parametri precedenti non è suff. nel caso in cui =1. In tale caso si variano diversi parametri tra cui la fase stazionaria, la fase mobile e la T.")
44
Scarsa selettività ed efficienza Aumento di efficienza ma
= picchi non risolti Aumento di efficienza Aumento di selettività Aumento di efficienza ma non di selettività

45
Il problema generale della eluizione

46
Eluizione isocratica:
Composizione fase mobile costante HPLC Eluizione a gradiente: Composizione fase mobile variabile nel tempo Condizione isoterma: Temperatura costante GC Programmazione di temperatura: Temperatura variabile

47
Asimmetria dei picchi cromatografici
I picchi cromatografici possono avere una forma gaussiana non simmetrica. Si possono avere due tipi di deformazioni: tailing: il tracciato sale bruscamente per scendere lentamente fronting: il tracciato sale lentamente e scende rapidamente

48
Il fattore di simmetria viene espresso dal fattore o rapporto di asimmetria:
As= b/a Ove a e b sono le distanze della curva dalla verticale tracciata nel punto di massimo del picco, misurate in corrispondenza del 10% dell’altezza del picco, rispettivamente a sinistra e destra del punto massimo. Nel caso di tailing As>1 mentre nel caso di fronting As<1.

49
Le cause di fronting e tailing possono essere ricondotte ad una di queste cause:
Introduzione del campione lenta o comunque scorretta Adsorbimento irreversibile della sostanza nella fase stazionaria sovraccarico Ridotta solubilità del campione nella fase mobile 2 mg 2 µg

Presentazioni simili
















>")
 Isolamento e Purificazione dei Composti Organici>")

