Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
1 test (che riguarda la parte spiegata da me) il 4 dicembre.
Se vogliono farlo prima, proponi il 21 novembre

2
MALATTIE GENETICHE Mutazioni: genomiche, cromosomiche, geniche
Malattie da Imprinting Malattie genetiche da singolo gene ereditate in modo mendeliano Malattie da singolo gene non ereditate in modo mendeliano (Mitocondriali e Malattie da espansione di triplette) Tecniche diagnostiche
 Tecniche diagnostiche.")
3
MALATTIE GENETICHE Mutazioni Genomiche Mutazioni Cromosomiche
(n° Cromosomi # 46) Cromosomi Autosomici Cromosomi Sessuali NON EREDITARIE Mutazioni Cromosomiche (Alterazione Struttura Cromosomi) Traslocazioni Delezioni EREDITARIE E NON EREDITARIE Mutazioni Geniche Delezioni, Inserzioni, Mutazioni Puntiformi, Espansione di Triplette ) Possono essere EREDITATE in modo : MENDELIANO: singolo gene dominate/recessivo, cromosomi sessuali NON MENDELIANO: Espansione triplette, mitocondriali
 Cromosomi Autosomici. Cromosomi Sessuali. NON EREDITARIE. Mutazioni Cromosomiche. (Alterazione Struttura Cromosomi) Traslocazioni. Delezioni. EREDITARIE E NON EREDITARIE. Mutazioni Geniche. Delezioni, Inserzioni, Mutazioni Puntiformi, Espansione di Triplette ) Possono essere EREDITATE in modo : MENDELIANO: singolo gene dominate/recessivo, cromosomi sessuali. NON MENDELIANO: Espansione triplette, mitocondriali.")
4
PATOLOGIA GENETICA Differenza tra: Malattie genetiche
Studia i fenomeni patologici che riconoscono come causa un’alterazione del genoma Differenza tra: Malattie genetiche Malattie ereditarie Malattie congenite

5
Malattie genetiche: comprendono tutte quelle condizioni patologiche
a carico del patrimonio genetico (ereditarie e non) Malattie ereditarie: Derivano dai genitori, sono trasmesse attraverso le cellule germinali nelle diverse generazioni e sono quindi familiari Non tutte le malattie ereditarie si manifestano al momento della nascita (ex. Corea di Huntington) Malattie congenite: significa “Nato con”. I sintomi sono riscontrabili al momento della nascita. Non tutte le malattie congenite sono ereditarie (ex. Sindrome di Down, raramente lo e’) oppure determinate geneticamente (ex. Toxoplasmosi). Derivano da fattori patogeni di natura fisica/chimica/biologica che agendo durante la vita intrauterina inducono alterazioni organiche
 Malattie ereditarie: Derivano dai genitori, sono trasmesse attraverso le cellule germinali nelle diverse generazioni e sono quindi familiari. Non tutte le malattie ereditarie si manifestano al momento della nascita (ex. Corea di Huntington) Malattie congenite: significa Nato con . I sintomi sono riscontrabili. al momento della nascita. Non tutte le malattie congenite sono. ereditarie (ex. Sindrome di Down, raramente lo e’) oppure determinate. geneticamente (ex. Toxoplasmosi). Derivano da fattori patogeni di natura fisica/chimica/biologica che. agendo durante la vita intrauterina inducono alterazioni organiche.")
6
Mutazioni ed Evoluzione
L’insorgenza di mutazioni é dovuta ad una interazione fra componenti ambientali (esempio: radiazioni, raggi cosmici, composti chimici) e una non perfetta efficienza dei sistemi di riparazione dei danni al DNA Le mutazioni peró possono insorgere anche spontaneamente: una classe di DNA polimerasi é detta “a bassa fedeltá” proprio per la sua tendenza ad inserire errori: vi é pressione selettiva per la riduzione, non per la scomparsa delle mutazioni
 e una non perfetta efficienza dei sistemi di riparazione dei danni al DNA. Le mutazioni peró possono insorgere anche spontaneamente: una classe di DNA polimerasi é detta a bassa fedeltá proprio per la sua tendenza ad inserire errori: vi é pressione selettiva per la riduzione, non per la scomparsa delle mutazioni.")
7
NON TUTTE LE MUTAZIONI RISULTANO ESSERE CAUSA DI MALATTIE…
HIV-1 e DCCR5

8
Meccanismo di Infezione da HIV-1

9
Il Recettore CCR5 (CD195) Localizzato sul cromosoma 3
Recettore per le chemiochine RANTES, Mp1alfa e Mip1 beta Espresso su linfociti T, Macrofagi, cellule dendritiche e microgliali Esiste una mutazione D32 (32 nucleotidi) che produce una proteina non capace di raggiungere la superficie cellulare L’allele D32 è comune nella popolazione Caucasica (frequeunza 10%) ma quasi assente in Africa e Asia I portatori Omozigoti di questa mutazione non hanno CCR5 sulla superficie cellulare Sono resistenti all’infezione da HIV-1 (R5) Hanno un fenotipo normale
 che produce una proteina. non capace di raggiungere la superficie cellulare. L’allele D32 è comune nella popolazione Caucasica (frequeunza 10%) ma quasi assente in Africa e Asia. I portatori Omozigoti di questa mutazione non hanno CCR5. sulla superficie cellulare. Sono resistenti all’infezione da HIV-1 (R5) Hanno un fenotipo normale.")
10
Individui Recanti la Mutazione D32 sono protetti
dall’Infezione HIV-1 (R5)
")
11
Le Malattie Genetiche Sono Molto Frequenti
Nel corso della vita: 700 per ogni 1000 individui Il 50% degli aborti spontanei sono riconducibili ad alterazioni geniche Circa 1% di nuovi nati ha alterazioni cromosomiche Il 5 % di persone al di sotto dei 25 anni sviluppano malattie riconducibili a disordini genetici Sono compresi però sia disordini genetici classici (cioè ereditari) sia altre malattie riconducibili a disordini genetici ex.: cancro (genetiche ed ambientali) malattie cardiovascolari (genetiche e ambientali) Queste sono le principali cause di morte nel mondo occidentale
 sia altre malattie riconducibili a disordini genetici ex.: cancro (genetiche ed ambientali) malattie cardiovascolari (genetiche e ambientali) Queste sono le principali cause di morte nel mondo occidentale.")
12
Malattie Genetiche In funzione dell’estensione della mutazione posso essere suddivise in: Mutazioni genomiche (ANEUPLOIDIA): perdita o acquisto di interi cromosomi (ex. Sindrome di Down) 2) Mutazioni cromosomiche (riarrangiamento di materiale genomico, ex. traslocazioni) Definite anche come: Aberrazioni Cromosomiche 3) Mutazioni Geniche
: perdita o acquisto di interi cromosomi (ex. Sindrome di Down) 2) Mutazioni cromosomiche (riarrangiamento di materiale genomico, ex. traslocazioni) Definite anche come: Aberrazioni Cromosomiche. 3) Mutazioni Geniche.")
13
Aberrazioni Cromosomiche
Mutazioni genomiche (Aneuploidia) 2) Mutazioni cromosomiche (ex. Traslocazioni)
 2) Mutazioni cromosomiche (ex. Traslocazioni)")
14
Mutazioni genomiche Trisomia: 2n+1 Monosomia: 2n-1
A carico dei cromosomi autosomici sono rare Più frequenti a livello dei cromosomi sessuali

15
Mutazioni genomiche (Cause)
Alterazione della meiosi durante la gametogenesi Alterazioni mitotiche durante le prime fasi dello sviluppo (MOSAICISMO) (46,XY/47,XY,+21) In entrambi i casi il fenomeno più frequente è di: NON DISGIUNZIONE (meiotica/mitotica) Raramente SEGREGAZIONE (ritardo nella migrazione del cromosoma)
 (46,XY/47,XY,+21) In entrambi i casi il fenomeno più frequente è di: NON DISGIUNZIONE (meiotica/mitotica) Raramente SEGREGAZIONE (ritardo nella migrazione del cromosoma)")
16
NON-DISGIUNZIONE: incapacità dei cromatidi fratelli appaiati di separarsi nella seconda divisione meiotica. I due cromosomi o cromatidi congiunti migrano ad un polo e vengono inclusi in una sola cellula figlia, mentre l’altra avrà materiale genetico in meno RITARDO ANAFASICO: ritardata migrazione del cromosoma durante l’anafase, conseguente perdita del cromosoma. Mancata incorporazione di un cromosoma nel nucleo di una delle cellule figlie.

17
Meiosi: è un processo di divisione mediante il quale una cellula eucariotica con corredo cromosomico diploide dà origine a quattro cellule con corredo cromosomico aploide. I Divisione meiotica II Divisione meiotica

18
(Cromosomi Autosomici)
Mutazioni genomiche (Cromosomi Autosomici) Sindrome di Down: trisomia 21 (incidenza 1/700) causata prevalentemente da non disgiunzione meiotica, a carico dell’ oocita aumenta con l’eta’ della madre (over 35 amniocentesi) 1/1550 sotto 20 anni, 1/25 sopra i 45 anni Più raramente (3-4% casi) causata da non disgiunzione mitotica in una fase più o meno precoce dello zigote (Mosaicismo) con fenotipo più lieve del precedente Traslocazione del braccio lungo del cromosoma 21 sul 14 (rara) (QUESTA E’ EREDITARIA)
 Sindrome di Down: trisomia 21 (incidenza 1/700) causata prevalentemente da non disgiunzione meiotica, a carico. dell’ oocita. aumenta con l’eta’ della madre (over 35 amniocentesi) 1/1550 sotto 20 anni, 1/25 sopra i 45 anni. Più raramente (3-4% casi) causata da non disgiunzione mitotica in. una fase più o meno precoce dello zigote (Mosaicismo) con fenotipo. più lieve del precedente. Traslocazione del braccio lungo del cromosoma 21 sul 14. (rara) (QUESTA E’ EREDITARIA)")
19
Sindrome di Down

20
Sindrome di Down La SD si associa sovente a complicanze malformative che richiedono interventi chirurgici rilevanti nel corso dei primi anni di vita: il 50% presenta malformazioni cardiache, il 30% stenosi duodenale, l’1% atresia esofagea, il 2% malformazioni anorettali. La chirurgia oftalmica è richiesta nel 12% dei casi per problemi di cataratta. Oltre alle malformazioni congenite descritte, il soggetto con SD ha la tendenza a sviluppare patologie secondarie per deficit nel sistema immunitario con particolare predisposizione ad infezioni batteriche; nell’1% poi dei casi compare leucemia acuta. Nel corso della vita il soggetto Down tende anche a sviluppare ipotiroidismo e diabete mellito.

21
(Cromosomi Autosomici)
Mutazioni genomiche (Cromosomi Autosomici) Edwards Syndrome: trisomia 18 (1/8000).The syndrome has a very low rate of survival, resulting from heart abnormalities, kidney malformations, and other internal organ disorders
 Edwards Syndrome: trisomia 18 (1/8000).The syndrome. has a very low rate of survival, resulting from heart abnormalities, kidney malformations, and other internal organ disorders.")
22
(Cromosomi Autosomici)
Mutazioni genomiche (Cromosomi Autosomici) Patau Syndrome: trisomia 13 (1/15000) common abnormalites include: Nervous system Musculoskeletal and cutaneous Urogenital Derivano quasi sempre da non disgiunzione meiotica nell’oocita. La probabilità Incrementa con l’età della madre A causa della severità del fenotipo, in entrambi i casi, la morte sopraggiunge nel primo anno di età
 Patau Syndrome: trisomia 13 (1/15000) common abnormalites include: Nervous system Musculoskeletal. and cutaneous Urogenital. Derivano quasi sempre da non disgiunzione meiotica nell’oocita. La probabilità Incrementa con l’età della madre. A causa della severità del fenotipo, in entrambi i casi, la morte. sopraggiunge nel primo anno di età.")
23
Mutazioni genomiche (Cromosomi Sessuali)
Più numerose perchè meglio tollerate: inattivazione di tutti i cromosomi X in eccesso tranne uno scarsa quantità di informazione genica presente sul cromosoma Y Tutte le malattie: Causano problemi di sviluppo sessuale e fertilità Sono normalmente evidenziabili durante la pubertà Danno ritardo mentale

24
Mutazioni genomiche (Cromosomi Sessuali)
Lyonization: (Lyon 1961) solo uno dei due cromosomi X rimane attivo (in modo casuale paterno/materno), l’altro viene represso durante le prime fasi dell’embriogenesi e la repressione persiste nella progenie. Femmine hanno mosaicismo derivante da cellule che hanno attivo il cromosoma X da parte del padre o da parte della madre . Evidenziabile dal corpo di Barr nei nuclei in interfase (massa al lato della membrana nucleare) - Oggi si sa che non tutti i geni del X inattivo sono veramente inattivi (ex. Sindrome di Turner femmine con un solo X) hanno severe anomalie somatiche e sessuali
 solo uno dei due cromosomi X rimane attivo (in modo casuale paterno/materno), l’altro viene represso durante le prime fasi dell’embriogenesi e la repressione persiste nella progenie. Femmine hanno mosaicismo derivante da cellule che hanno attivo il cromosoma X da parte del padre o da parte della madre . Evidenziabile dal corpo di Barr nei nuclei in interfase (massa al lato della membrana nucleare) - Oggi si sa che non tutti i geni del X inattivo sono veramente inattivi (ex. Sindrome di Turner femmine con un solo X) hanno severe anomalie somatiche e sessuali.")
25
Il gene deputato a tale processo è XIST (X inactive specific transcript), che trascrive un RNA espresso solo dal cromosoma inattivato e che non codifica alcuna proteina. Agisce sul centro di inattivazione dell'X (Xic, X inactivation center). XIST non è espresso nel maschio normale (46,XY). Il cromosoma X differisce dagli autosomi per alcune caratteristiche: geni corti, minor numero di geni, bassa densità di geni.

26
The X-inactive specific transcript (Xist) gene encodes a large non-coding RNA that is responsible for mediating the specific silencing of the X chromosome from which it is transcribed. The inactive X chromosome is coated by Xist RNA, whereas the Xa is not . The Xist gene is the only gene which is expressed from the Xi but not from the Xa. X chromosomes which lack the Xist gene cannot be inactivated. Artificially placing and expressing the Xist gene on another chromosome leads to silencing of that chromosome. Compared to the Xa, the Xi has high levels of DNA methylation, low levels of histone acetylation, low levels of histone H3 lysine-4 methylation, and high levels of histone H3 lysine-9 methylation, all of which are associated with gene silencing. The existence of genes along the inactive X which are not silenced explains the defects in humans with abnormal numbers of the X chromosome, such as Turner syndrome (X0) or Klinefelter syndrome (XXY).
 or Klinefelter syndrome (XXY).")
27
Esempi di malattie causate da anomalie
nel numero dei cromosomi sessuali Klinefelter syndrome: 47,XXY (1/1000) Turner syndrome: 45,X (1/5000) Multi-X Females: 47,XXX; 49,XXXXX XYY Sindrome: 47,XYY (1/1000) Un solo cromosoma Y e’ in grado di determinare il sesso, su di esso e’ presente il gene SRY (Sex-determining Region Y) che determina la soppressione dei caratteri primari femminili (utero, tube e ovaio) e promuove lo sviluppo dei caratteri sessuali maschili Codifica per un fattore trascrizionale (HMG-box family) che inizia la determinazione sessuale maschile
 Turner syndrome: 45,X (1/5000) Multi-X Females: 47,XXX; 49,XXXXX. XYY Sindrome: 47,XYY (1/1000) Un solo cromosoma Y e’ in grado di determinare il sesso, su di esso. e’ presente il gene SRY (Sex-determining Region Y) che determina la. soppressione dei caratteri primari femminili (utero, tube e ovaio) e promuove. lo sviluppo dei caratteri sessuali maschili. Codifica per un fattore trascrizionale (HMG-box family) che. inizia la determinazione sessuale maschile.")
28
Klinefelter syndrome: 47,XXY (1/1000)
")
29
Klinefelter syndrome: 47,XXY (1/1000)
mancato sviluppo dei caratteri sessuali secondari microrchidia e aspermatogenesi tendenza all'alta statura L'analisi dei cromosomi sui linfociti è lo standard genetico di diagnosi deficit di androgeni solo il 10% presenta un ritardo mentale. ridotto sviluppo del linguaggio, con problemi di espressività sul piano comportamentale si possono riscontrare immaturità, poca sicurezza, timidezza

30
Turner syndrome: 45,X (1/5000) Sintomi della sindrome di Turner sono:
ipogonadismo con fenotipo femminile bassa statura torace a scudo (piatto) e capezzoli iperdistanziati; attaccatura dei capelli bassa orecchie a basso impianto; il viso può avere un aspetto da persona anziana sterilità dovuta a malformazioni dell'ovaia detta "a stria"; amenorrea primaria, cioè, l'assenza della mestruazione (in Turner la menopausa avviene prima del menarca all'età di 2 anni);
 e capezzoli iperdistanziati; attaccatura dei capelli bassa. orecchie a basso impianto; il viso può avere un aspetto da persona anziana. sterilità dovuta a malformazioni dell ovaia detta a stria ; amenorrea primaria, cioè, l assenza della mestruazione (in Turner la menopausa avviene prima del menarca all età di 2 anni);")
31
Multi-X Females: 47,XXX; 49,XXXXX
Le principali caratteristiche standard sono: altezza (maggiore che nella media) circonferenza cranica inferiore alla norma (soprattutto alla nascita, poi c’è un ridotto recupero). sviluppo motorio generalmente un po’ ritardato. Inizio della pubertà tendenzialmente ritardato. sviluppo cognitivo. In media di poco inferiore alla norma. sviluppo comunicativo e linguistico. Tendenzialmente allo stesso livello o inferiore rispetto alle prestazioni intellettive generali.
 circonferenza cranica inferiore alla norma (soprattutto alla nascita, poi c’è un ridotto recupero). sviluppo motorio generalmente un po’ ritardato. Inizio della pubertà tendenzialmente ritardato. sviluppo cognitivo. In media di poco inferiore alla norma. sviluppo comunicativo e linguistico. Tendenzialmente allo stesso livello o inferiore rispetto alle prestazioni intellettive generali.")
32
XYY Sindrome: 47,XYY (1/1000) I soggetti possono presentare spesso un'altezza superiore a 180 cm (media 188 cm)
 I soggetti possono presentare spesso un altezza superiore a 180 cm (media 188 cm)")
33
Mutazioni cromosomiche
(Anomalie strutturali dei cromosomi ex. Traslocazioni/Delezioni)
")
34
Mutazioni cromosomiche
Rottura Perdita di un braccio e duplicazione di quello rimanente Rottura e fusione delle estremita’ problemi in meiosi/mitosi

35
LEUCEMIA PROMIELOCITICA ACUTA TRASLOCAZIONE BILANCIATA t (15;17)
")
36
Mutazioni cromosomiche

37
TRASLOCAZIONE ROBERTSONIANA
Not Viable La persona portatrice di questa anomalia è perfettamente sana. È da considerare, però, che un’alta percentuale dei gameti prodotti dalle persone con traslocazioni bilanciate/ robertsoniane può essere “sbilanciata”. Di conseguenza, le persone che hanno una traslocazione, nella loro vita riproduttiva, hanno un rischio superiore, rispetto a quello di chi non è portatore di traslocazioni, di avere figli con patologia malformativa o di avere gravidanze interrotte da aborti

38
(Prader-Willi, Angelman)
Malattie da Genomic Imprinting (Prader-Willi, Angelman)
")
39
Genomic imprinting Per imprinting genomico si intende una modificazione “epigenetica” di uno specifico allele nel gamete o nello zigote, responsabile per l’espressione differenziale dei due alleli del gene nelle cellule somatiche della progenie For the vast majority of autosomal genes, expression occurs from both alleles simultaneously. In mammals however, a small proportion (<1%) of genes are imprinted, meaning that gene expression occurs from only one allele.The expressed allele is dependent upon its parental origin. For example, the gene encoding Insulin-like growth factor 2 (Igf2) is only expressed from the allele inherited from the father.
 of genes are imprinted, meaning that gene expression occurs from only one allele.The expressed allele is dependent upon its parental origin. For example, the gene encoding Insulin-like growth factor 2 (Igf2) is only expressed from the allele inherited from the father.")
40
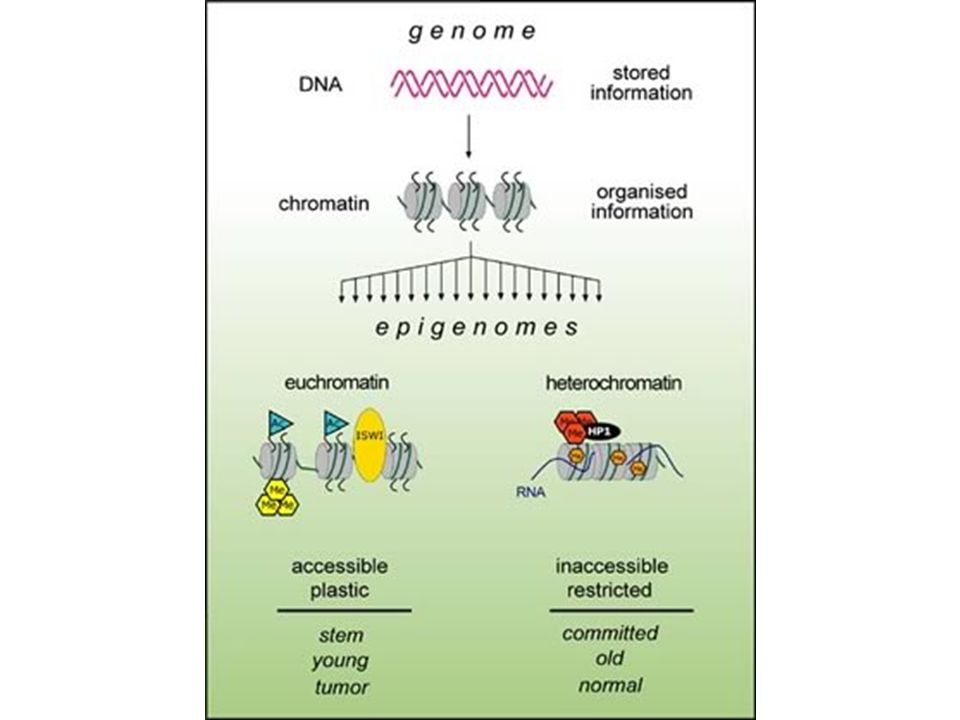
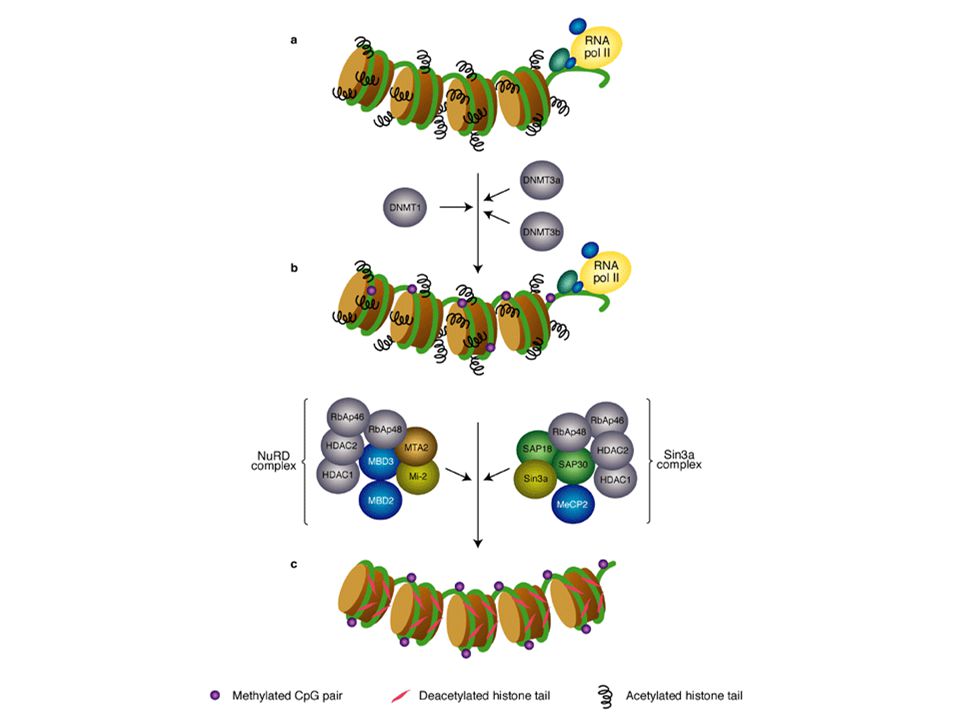
EPIGENETICA Per modificazione epigenetica, si intende una modificazione ereditaria genomica, che non é associata ad un cambio della sequenza di DNA.

44
Imprinting control region
Demetilated maternal region La delezione di ICR porta all’espressione materna di igf2

45
Malattie da Imprinting
Prader-Willi syndrome Angelman syndrome

46
Prader-Willi Syndrome
E’ una malattia genetica rara (colpisce 1 su nati vivi) caratterizzata dall'alterazione del cromosoma 15 Ipotonia nell' infanzia, obesità dovuta ad iperfagia, ipogonadismo, mani e piedi piccoli E’ la più comune tra le sindromi di microdelezione cromosomica Avviene per due diverse cause accertate, entrambe di tipo genetico: Delezione sul cromosoma 15 paterno Disomia uniparentale materna del cromosoma 15
 caratterizzata dall alterazione del cromosoma 15. Ipotonia nell infanzia, obesità dovuta ad iperfagia, ipogonadismo, mani e piedi piccoli. E’ la più comune tra le sindromi di microdelezione cromosomica. Avviene per due diverse cause accertate, entrambe di tipo genetico: Delezione sul cromosoma 15 paterno. Disomia uniparentale materna del cromosoma 15.")
47
Prader-Willi/Angelman Syndrome
Nella PWS il gene materno è silenziato perché sotto imprinting, mentre quello paterno è deleto La regione in questione è sul cromosoma 15 (15q11-q13). La PWS è strettamente correlata con la Sindrome di Angelman (AS), che è causata da imprinting paterno e delezione del gene materno caratterizzata da movimenti ripetitivi, simmetrici, atassici e da una disposizione all' allegria, al riso frequente
. La PWS è strettamente correlata con la Sindrome di Angelman (AS), che è causata da imprinting paterno e delezione del gene materno. caratterizzata da movimenti ripetitivi, simmetrici, atassici e da una. disposizione all allegria, al riso frequente.")
48
Stage 1 In the first stage, infants with PWS are hypotonic or "floppy", with very low muscle tone. Weak cry and a poor suck reflex are typical. Babies with PWS usually are unable to breastfeed and frequently require tube feeding. These infants may suffer from "failure to thrive" if feeding difficulties are not carefully monitored and treated. As these children grow older, strength and muscle tone generally improve. Motor milestones are achieved, but are usually delayed. Stage 2 An unregulated appetite characterizes the second stage of PWS. This stage most commonly begins between ages 2 and 6 years old. Individuals with PWS lack normal hunger and satiety cues. They usually are not able to control their food intake and will overeat if not closely monitored. Food seeking behaviors are very common. In addition, the metabolic rate of persons with PWS is lower than normal. Left untreated, the combination of these problems will lead to morbid obesity and its many complications

49
(Prader-Willi and Angelman Syndromes)
Genomic Imprinting (Prader-Willi and Angelman Syndromes) Sono attivi solo: Un set di geni Prader-Willi (paterni) Un set di geni Angelman (materni) Delezione banda q12 Cromosoma 15 Uniparental Disomy Stesso risultato delle delezione Sono attivi solo: Un set di geni Prader-Willi (paterni) Sono attivi solo: Un set di geni Angelman (materni)
 Sono attivi solo: Un set di geni Prader-Willi (paterni) Un set di geni Angelman (materni) Delezione banda q12. Cromosoma 15. Uniparental Disomy. Stesso risultato delle. delezione. Sono attivi solo: Un set di geni Prader-Willi (paterni) Sono attivi solo: Un set di geni Angelman (materni)")
50
Uniparental Disomy Perdita del cromosoma Materno (PWS) o
Paterno (Angelman S)
")
51
MUTAZIONI GENICHE EREDITARIE
Tutte quelle malattie derivanti da alterazioni genetiche che: possono essere trasmesse per via parentale perché presenti nelle cellule germinali Normalmente: non alterano la capacità riproduttiva sono compatibili con la vita

52
DEFINIZIONE DI MUTAZIONE
Permanente cambio nella sequenza del DNA Tutte le mutazioni che avvengo a livello delle cellule germinali possono essere trasmesse alla progenie e dare luogo a malattie ereditarie Le mutazioni a carico di cellule somatiche non vengono trasmesse per via ereditaria ma sono importanti nello sviluppo di cancro e malattie congenite

53
Tipi di Mutazioni Geniche (I)
Possono verificarsi nelle regioni: codificanti, regolatorie (ex promotori) e nei siti di splicing Mutazioni puntiformi: sostituzione di aa che causano cambio di sequenza o stop codon (talassemia) Frame shift: delezione o inserzione di nucleotidi (cambio intera sequenza aa a valle della mutazione) Inserzione/delezione di triplette: aggiunta o eliminazione di aa Espansione di triplette: caratterizzate dall’essere dinamiche perchè aumentano durante la gametogenesi (ex.X-Fragile e Corea Huntington)
 e nei siti di splicing. Mutazioni puntiformi: sostituzione di aa che causano cambio di sequenza o stop codon (talassemia) Frame shift: delezione o inserzione di nucleotidi (cambio intera sequenza aa a valle della mutazione) Inserzione/delezione di triplette: aggiunta o eliminazione di aa. Espansione di triplette: caratterizzate dall’essere dinamiche perchè aumentano durante la gametogenesi (ex.X-Fragile e Corea Huntington)")
54
Mutazioni puntiformi Introduzione stop Autosomica recessiva
beta° talasemia

55
Mutazioni “frame-shift”
Cambio della sequenza aa a valle della mutazione

56
Delezioni di triplette
Eliminazione di aa con alterazione della struttura/funzione della proteina)
")
57
Espansione di triplette

58
Classificazione delle malattie ereditarie
Mendeliane (mutazione di singoli geni con ampio effetto) Multifattoriali (genetici e ambientali) Malattie da singolo gene con trasmissione non mendeliana (ex. espansione da triplette, malattie mitocondriali)
 Multifattoriali (genetici e ambientali) Malattie da singolo gene con trasmissione non mendeliana (ex. espansione da triplette, malattie mitocondriali)")
59
(mutazione di singoli geni con ampio effetto)
Mendeliane (mutazione di singoli geni con ampio effetto) Autosomiche domaninanti (ex. Ipercolesterolemia Familiare) Autosomiche recessive (ex. Fibrosi cistica) Sex-Linked (ex. Emofilia A)
 Autosomiche domaninanti (ex. Ipercolesterolemia Familiare) Autosomiche recessive (ex. Fibrosi cistica) Sex-Linked (ex. Emofilia A)")
60
Autosomiche Dominanti (I)
Manifeste clinicamente allo stato eterozigote (maschi e femmine affetti in egual misura) - Fenotipo influenzato da: penetranza (% di individui che, avendo il gene malato, manifesta il fenotipo) espressivitá (livello di fenotipo) Dovute alla presenza di geni modificanti
 - Fenotipo influenzato da: penetranza (% di individui che, avendo il gene malato, manifesta il fenotipo) espressivitá (livello di fenotipo) Dovute alla presenza di geni modificanti.")
61
Autosomiche Dominanti (II)
Normalmente coinvolgono DUE TIPI DI MUTAZIONI: Mutazione per Perdita di funzione Mutazione per Guadagno di funzione E DUE CATEGORIE DI PROTEINE Proteine regolatorie (ex. LDLr Familial Hypercholesterolemia), - Proteine strutturali (ex. Collagene in Osteogenisis Imperfecta )
, - Proteine strutturali (ex. Collagene in Osteogenisis Imperfecta )")
62
Pedigree di una Malattia
Autosomica Dominante

64
Autosomiche domaninanti (ex. Familial Hypercholesterolemia)
E’ una malattia recettoriale causata da una mutazione nel gene codificante: Il recettore per Low-density lipoprotein (LDL) coinvolto nel trasporto e metabolismo del colesterolo che causa un elevato livello di colesterolo nel plasma (2-3 volte rispetto alla media) Caratteristiche soggetti affetti (1/500): precoci lesioni aterosclerotiche infarto e/o ictus cerebrale con insorgenza giovanile Xantomi (accumuli di grasso) tendinei e cutanei
 coinvolto nel. trasporto e metabolismo del colesterolo che causa un elevato. livello di colesterolo nel plasma (2-3 volte rispetto alla media) Caratteristiche soggetti affetti (1/500): precoci lesioni aterosclerotiche. infarto e/o ictus cerebrale con insorgenza giovanile. Xantomi (accumuli di grasso) tendinei e cutanei.")
65
Metabolismo delle LDL Adipociti e muscoli Estrazione trigliceridi 2nd
Ricche in trigliceridi Estrazione trigliceridi 1st 4th Tramite LDLr 70% dal fegato Restante da Fibroblasti, linfociti, muscolatura liscia 3rd 5th Aumentata nei malati perche’ IDL non assorbiti

66
Il metabolismo del colesterolo

67
CLASSIFICAZIONE DELLE MUATAZIONI LDLr
BASATE SULLE FUNZIONI DELLE PROTEINE MUTATE Il gene per LDLr è situato sul cromosoma 19, comprende 18 esoni e 5 domini. Sono state mappate almeno 150 mutazioni Che differentemente modificano la proteina

68
Autosomiche Recessive (I)
Entrambi i geni devono essere mutati L’espressione e’ piu’ uniforme rispetto alle malattie autosomiche dominanti Penetranza completa e’ comune In molti casi proteine enzimatiche sono affette da perdita di funzione Pedigree di un carattere recessivo Consanguinei

70
Autosomiche Recessive (III)
(ex. FIBROSI CISTICA) Difetto del trasporto del cloro a livello epiteliale causato da mutazioni nel gene CFTR (codificante per il canale dello ione cloro) Codifica una proteina di 1480 aminoacidi situata sulla membrana cellulare delle cellule epiteliali, la cui funzione, normalmente, è quella di trasportare il cloro attraverso le membrane cellulari a livello della membrana apicale delle cellule epiteliali delle cellule di vie aeree, del pancreas, dell'intestino, delle ghiandole sudoripare, delle ghiandole salivari e dei vasi defereni Insorgenza 1/4000
 Difetto del trasporto del cloro a livello epiteliale. causato da mutazioni nel gene CFTR (codificante per il canale dello ione cloro) Codifica una proteina di 1480 aminoacidi situata sulla membrana cellulare delle cellule. epiteliali, la cui funzione, normalmente, è quella di trasportare il cloro attraverso le. membrane cellulari a livello della membrana apicale delle cellule epiteliali delle cellule. di vie aeree, del pancreas, dell intestino, delle ghiandole sudoripare, delle. ghiandole salivari e dei vasi defereni. Insorgenza 1/4000.")
71
CARATTERISTICHE Negli organi interessati, le secrezioni mucose, essendo anormalmente viscide, determinano un'ostruzione dei dotti principali, provocando l'insorgenza di gran parte delle manifestazioni cliniche tipiche della malattia Coinvolge: ghiandole esocrine, epiteli respiratori, intestinali e riproduttivi produzione di secrezioni mucose viscose che causano: Infezioni polmonari Insufficienza pancreatica Cirrosi epatica Infertilità maschile Occlusioni intestinali Mal nutrizione

72
Polmoni: Il quadro clinico è dominato da un lento processo distruttivo polmonare. Nella maggior parte dei casi si manifesta nel primo anno di vita con tosse persistente. L'esame obbiettivo può subito dimostrare segni indiretti di ostruzione bronchiale. L'infezione bronchiale cronica determina una progressiva distruzione del parenchima polmonare. La risposta immunitaria dell'ospite e i fattori propri dei patogeni contribuiscono quindi insieme ad innescare un processo patogenetico che è alla base del processo distruttivo polmonare. Pancreas: è colpito nell'80% dei casi con un ristagno dei succhi pancreatici nei dotti con formazione di cisti con una fibrosi che si va a creare attorno a questi (da qui fibrosi cistica). La carenza di succhi pancreatici nel canale intestinale porta a malassorbimento di grassi (con conseguente steatorrea), e di conseguenza delle vitamine liposolubili, delle proteine e, in minima parte, degli zuccheri. Con il passare del tempo il pancreas, sempre più colpito, secerne una minor quantità di insulina portando a una forma di diabete di solito insulino-dipendente.
. La carenza di succhi pancreatici nel canale intestinale porta a malassorbimento di grassi (con conseguente steatorrea), e di conseguenza delle vitamine liposolubili, delle proteine e, in minima parte, degli zuccheri. Con il passare del tempo il pancreas, sempre più colpito, secerne una minor quantità di insulina portando a una forma di diabete di solito insulino-dipendente.")
73
This higher-power photomicrograph of the pancreas shows interstitial tissue and the presence of small cystic spaces (1) within the acinar lobules. These spaces are filled with an eosinophilic proteinaceous material. The islets of Langerhans (2) are unaffected.
 are unaffected..")
74
CANALE DEL CLORO 2 Domini trasmembrana 2 NBD Regulatory domain
550 mutazioni diverse La piu’ comune è una delezione di tripletta codificante per phe508 (folding non corretto)
")
75
FIBROSI CISTICA ESPRESSIVITA’
Classe I: non c'è produzione di proteina Classe II: si ha produzione di un corto peptide non funzionante Classe III: si produce un polipetide non funzionante Classe IV: si produce una proteina difettosa ma in minima parte funzionante Classe V: si produce una proteina normale ma in minime quantità Classe WT: la proteina normale è prodotta nelle giuste quantità (soggetto sano)
")
76
Disfunzione Ghiandole Sudoripare
Ridotto assorbimento Na+ Cl- Il rischio di disidratazione, specialmente nel periodo estivo, è molto elevato Metodo di Gibson e Cooke - si misura la concentrazione di Cloro in almeno 75 mg di sudore Valori sono superiori alla norma (60mEq/L) il test è sicuramente positivo Valori sotto i 30mEq/L è sicuramente negativo
 il test è sicuramente positivo. Valori sotto i 30mEq/L è sicuramente negativo.")
77
Disfunzione Vie Aeree Superiori
Aumento assorbimento Na+/H2O

78
Sex-Linked Sono tutti X-Linked e per la maggior parte recessivi
Modalità di trasmissione caratteristica: - I maschi non trasmettono la malattia ai figli maschi (ma le figlie femmine sono portatrici) - Le femmine a causa dell’ inattivazione casuale del cromosoma x hanno un fenotipo variabile
 - Le femmine a causa dell’ inattivazione casuale del cromosoma x hanno un fenotipo variabile.")
79
X-Linked Recessivo (ex. Emofilia A)
Causata dalla riduzione della quantità o dell’attività del fattore VIII VIII fondamentale per l’attivazione del fattore X Fenotipo alcune volte riscontrato anche in femmine eterozigoti (causato dall’inattivazione del cromosoma X wt nella maggior parte delle cellule) La severità della malattia correla con il tipo di mutazione (più l’attività del fattore VIII e’ bassa maggiore e’ la severità del fenotipo)
 La severità della malattia correla. con il tipo di mutazione. (più l’attività del fattore VIII e’ bassa. maggiore e’ la severità del fenotipo)")
80
Malattie genetiche multifattoriali
Risultanti dall’interazione fra le condizioni ambientali e un numero di geni >1 partecipanti alla determinazione del fenotipo Il rischio di presentare un fenotipo “malattia” é proporzionale al numero di geni mutati ereditato Molte delle piú comuni malattie (diabete, ipertensione, celiachia) hanno origine multifattoriale
 hanno origine multifattoriale.")
81
IL DIABETE MELLITO Con questo termine sono comprese alcune malattie, differenti per eziologia, patogenesi e decorso clinico, ma aventi come denominatore comune il sintomo di iperglicemia persistente a digiuno (>180mg/dl) (normale mg/dl) Associata poliuria e glucosuria Viene suddiviso in primario e secondario Primario: diabete mellito insulino dipendente (IDDM), giovanile diabete mellito non insulino dipendente (NIDDM), adulto Secondario: Pancreasectomia Pancreatiti Assenza dei recettori per l’insulina Modificazioni strutturali dell’insulina Provocano notevoli alterazioni metaboliche che portano a complicanze a carico del sistema circolatorio, nervoso, reni e occhi
 (normale mg/dl) Associata poliuria e glucosuria. Viene suddiviso in primario e secondario. Primario: diabete mellito insulino dipendente (IDDM), giovanile. diabete mellito non insulino dipendente (NIDDM), adulto. Secondario: Pancreasectomia. Pancreatiti. Assenza dei recettori per l’insulina. Modificazioni strutturali dell’insulina. Provocano notevoli alterazioni metaboliche che portano a. complicanze a carico del sistema circolatorio, nervoso, reni e occhi.")
82
Pancreas Esocrino: Endocrino (Isole di Langerhans)
I principali enzimi prodotti dal succo pancreatico sono: tripsinogeno, chimotripsinogeno, elastasi, lipasi pancreatiche, amilasi pancreatiche, fosfolipasi pancreatica, nucleasi pancreatiche. Endocrino (Isole di Langerhans) Cellule A (15%): glucagone Cellule B (80%). Insulina Cellule D (3-5%): somatostatina Cellule F (3-5%): polipeptide pancreatico
 Cellule A (15%): glucagone. Cellule B (80%). Insulina. Cellule D (3-5%): somatostatina. Cellule F (3-5%): polipeptide pancreatico.")
83
OMEOSTASI GLUCIDICA Carboidrati alimentari Glucosio Fruttosio
Galattosio Sangue Intestino Fegato Glicogeno di riserva Lattato Amminoacidi Glicerolo Deposito di trigliceridi Glicogenolisi Neoglicogenesi Cellule dei vari tessuti

84
Glucosio nel sangue Elevata concentrazione Insulina Glucagone Bassa concentrazione Stimolazione Glicolisi Captazione cellulare/utilizzazione metabolica del glucosio Glicogenosintesi Sintesi proteica lipogenesi Inibizione catabolismo anabolismo Glicogenolisi Neoglicogenolisi Lipolisi Captazione cellulare del glucosio Neoglicogenesi Chetogenesi

85
ASPETTI FISIOPATOLOGICI DELLA SINDROME DIABETICA
Riduzione della penetrazione di glucosio nelle cellule: Deficit insulinico o recettoriale Aumento della glicemia al di sopra di 180mg/dl si ha glucosuria (perdita glucosio con le urine, aumento diuresi osmotica eccesiva perdita di liquidi) Riduzione contenuto proteico (eccessiva neoglicogenesi) Glicosilazione proteine (danno) Incremento della lipolisi: aumento AcCoA, corpi chetonici, chetonemia, acidosi metabolica, come diabetico Aterosclerosi: aumento trigliceridi, VLDL e riduzione HDL Macroangiopatia diabetica (arterie coronariche, cerebrali, estremita’) Microangiopatia diabetica (coinvolgimento delle arteriole del microcircolo)
 Riduzione contenuto proteico (eccessiva neoglicogenesi) Glicosilazione proteine (danno) Incremento della lipolisi: aumento AcCoA, corpi chetonici, chetonemia, acidosi metabolica, come diabetico. Aterosclerosi: aumento trigliceridi, VLDL e riduzione HDL. Macroangiopatia diabetica (arterie coronariche, cerebrali, estremita’) Microangiopatia diabetica (coinvolgimento delle arteriole del microcircolo)")
86
type 1 diabetes—an autoimmune disease in which the body's own immune system attacks the pancreas, rendering it unable to produce insulin Compartecipazione genetica : frequente familiarita’, associazione aplotipo HLA DR3/4 Compartecipazione ambientale: gemelli omozigotici malatta <50% dei casi Terapia :insulinica (ricombinante umana)
")
87
type 2 diabetes—in which a resistance to the effects of insulin or a defect in insulin secretion may be seen Rappresenta l’85% di tutte le forme di diabete Compartecipazione genetica : frequente familiarita’, forse recettori insulinici Compartecipazione ambientale: iperalimentazione/obesita’

Presentazioni simili