Scaricare la presentazione
1
Cenni di spettroscopia UV/VIS

2
La spettroscopia UV/VIS Tecnica spettroscopica basata sulle interazioni tra gli elettroni, solitamente di valenza, e la componente elettrica della radiazione elettromagnetica nel campo del visibile e dellultravioletto. UV vicino UV lontano UV-A: 400-320 nm UV-B: 320-280 nm UV-C: 280- 10 nm

3
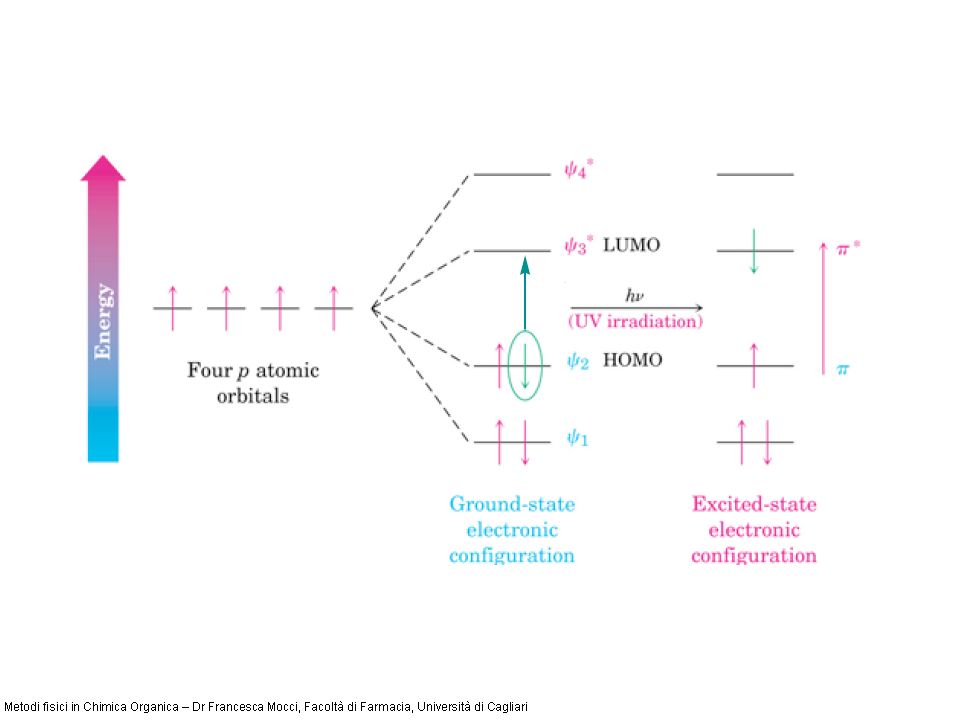
Le transizioni promosse dalla radiazione UV/Vis sono transizioni elettroniche dallo stato elettronico fondamentale ad uno stato elettronico eccitato ( spettroscopia elettronica). E Stato elettronico fondamentale Stato elettronico eccitato h E Normalmente le transizioni coinvolgono un solo elettrone per molecola, che generalmente si trova nellorbitale molecolare occupato a più alta energia (HOMO) e viene promosso allorbitale molecolare non occupato a più bassa energia (LUMO)
 e viene promosso allorbitale molecolare non occupato a più bassa energia (LUMO).")
4
4 orbitali atomici p

6
Livelli vibrazionali Livelli rotazionali Stato elettronico fondamentale Stato elettronico eccitato

7
La legge di Lambert e Beer I0I0 Campione ItIt A Rivelatore

8
La legge di Lambert e Beer I0I0 Campione ItIt Rivelatore A Cammino ottico. Lunghezza, l Concentrazione, c

9
La legge di Lambert e Beer I0I0 Campione ItIt Rivelatore A Cammino ottico. Lunghezza, l Concentrazione, c Assorbanza (adimensionale) Coefficiente di estinzione molare, 1000*cm 2 mol -1 Concentrazione, mol*l -1 Cammino ottico, cm
 Coefficiente di estinzione molare, 1000*cm 2 mol -1 Concentrazione, mol*l -1 Cammino ottico, cm.")
10
La legge di Lambert e Beer La legge di Lambert-Beer descrive bene il comportamento in assorbimento di soluzioni diluite, cioè con concentrazione generalmente fino a 0,01 M. Assorbanza (adimensionale) Coefficiente di estinzione molare, 1000*cm 2 mol -1 Concentrazione, mol*l -1 Cammino ottico, cm
 Coefficiente di estinzione molare, 1000*cm 2 mol -1 Concentrazione, mol*l -1 Cammino ottico, cm.")
11
Regole di selezione Affinchè una radiazione possa essere assorbita occorre che la sua energia (h ) sia uguale alla alla separazione tra due livelli energetici; questa è una condizione necessaria ma non sufficiente. Infatti occorre anche che le funzioni donda che descrivono gli stati iniziali e finali della transizione obbediscano a certi requisiti: le funzioni donda devono avere diversa simmetria gli stati di partenza e di arrivo devono avere la stessa molteplicità di spin (Molteplicità = 2s+1, s: spin totale) sono proibite le transizioni tra livelli energetici coinvolgenti più di un elettrone
 sono proibite le transizioni tra livelli energetici coinvolgenti più di un elettrone.")
12
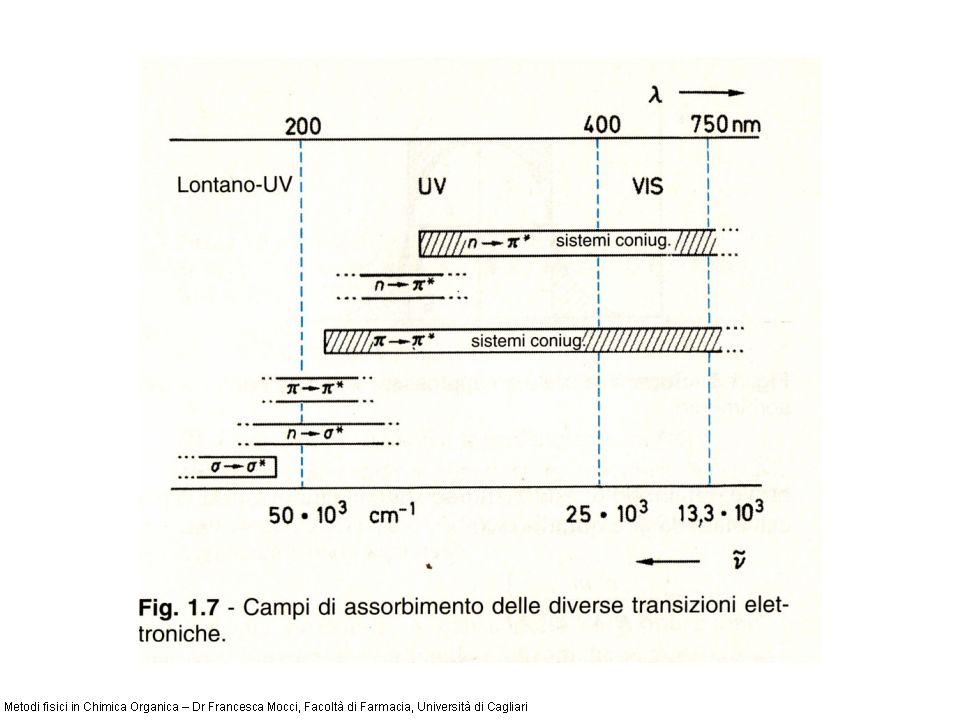
Classificazione delle transizioni elettroniche Le transizioni elettroniche possono essere classificate sulla base delle caratteristiche degli orbitali molecolari coinvolti. Le principali transizioni elettroniche, che coinvolgono elettroni di legame e lone-pair in una molecola organica, sono riassunte nello schema seguente n legame anti-legame non-legame

13
Le transizioni che coinvolgono elettroni, in particolare le transizioni *, sono transizioni intense hanno in genere energia molto elevata e le bande relative cadono nel lontano UV, intorno ai 135 nm. Ad esempio il metano, che possiede solo legami C-H, può dar luogo solo a questo tipo di transizione mostra un massimo di assorbanza a 125 nm. Transizioni di questo tipo non sono visibili con i normali spettrofotometri e bisogna far ricorso a tecniche di registrazione in vuoto. Transizioni *

14
Sono transizioni intense. In olefine semplici cadono intorno ai 170- 200 nm per cui non sono normalmente rilevabili. Diverso è invece il discorso quando si ha un diene coniugato, poiché le diolefine assorbono in una regione accessibile ai normali spettrometri UV. Transizioni *

15
Transizioni n * Transizioni abbastanza intense che coinvolgono elettroni di non legame (doppietti elettronici) che perciò possono essere osservate solo in molecole contenenti eteroatomi come O, N, S, alogeni, etc. Derivati alchilici sostituiti assorbono a lunghezze donda maggiori dei corrispondenti idrocarburi saturi (ad es. il massimo del CH 3 OH cade a ca. 185 nm contro i ca. 125 del CH 4 ), tuttavia in genere hanno energia elevata e cadono nel lontano - inizio vicino UV.
, tuttavia in genere hanno energia elevata e cadono nel lontano - inizio vicino UV..")
16
Transizioni n * Transizioni molto deboli che possono essere osservate in molecole in cui i gruppi portanti gli eteroatomi sono insaturi (chetoni, enoleteri, etc) e cadono nel vicino UV.
 e cadono nel vicino UV.")
18
Terminologia UV-vis CROMOFORO: CROMOFORO: gruppo responsabile dellassorbimento della radiazione; nel vicino UV o nel visibile sono normalmente gruppi insaturi come il C=O, i fenili etc. La posizione ed intensità dei massimi dei cromofori può variare al variare della natura dei sostituenti o del solvente. AUXOCROMI: AUXOCROMI: gruppi che non assorbono di per se stessi in maniera particolarmente intensa ma che provocano una variazione del massimo di assorbimento e/o dell'intensità dell'assorbimento. Ad es. sono gruppi auxocromi -OH, SH, OR, SR, NR 2, Alogeni etc (in genere contengono un atomo con un doppietto elettronico di non legame).
..")
19
Terminologia UV-vis CROMOFORO: CROMOFORO: gruppo responsabile dellassorbimento della radiazione; nel vicino UV o nel visibile sono normalmente gruppi insaturi come il C=O, i fenili etc. La posizione ed intensità dei massimi dei cromofori può variare al variare della natura dei sostituenti o del solvente.

20
Terminologia UV-vis Lo spostamento del massimo di assorbimento è definito: Batocromico Batocromico quando avviene verso lunghezze donda maggiori (red shift, verso il rosso) Ipsocromico Ipsocromico quando avviene verso lunghezze donda minori (blue shift, verso il blu)
 Ipsocromico Ipsocromico quando avviene verso lunghezze donda minori (blue shift, verso il blu)")
21
Terminologia UV-vis Leffetto sullintensità della banda di assorbimento è definito: Ipercromico Ipercromico quando si ha aumento dellintensità Ipocromico Ipocromico quando si ha diminuzione dellintensità

22
I cromofori

23
Cromoforo etilenico Letilene presenta un massimo di assorbimento * a 170 nm. Nel butadiene e nel trans esatriene i corrispondenti assorbimenti si osservano a ca. 215 e 255 nm e aumentando il numero di doppi legami coniugati si osserva un progressivo spostamento verso lunghezze donda più elevate (il -carotene -11 doppi legami - assorbe nel visibile), Analogamente per effetto di sostituenti auxocromi contenenti elettroni n, si osserva un effetto batocromico (regole di Woodward, Fieser e Scott).
, Analogamente per effetto di sostituenti auxocromi contenenti elettroni n, si osserva un effetto batocromico (regole di Woodward, Fieser e Scott)..")
24
Cromoforo carbonilico In aldeidi e chetoni saturi le intense transizioni * sono localizzate nel lontano UV, mentre la transizione n *, di debole intensità, cade nellintervallo 275-230 nm. Gruppi auxocromi legati direttamente al carbonile spostano la transizione n * verso lunghezze donda più corte (effetto ipsocromico). Transizioni n * in composti carbonilici saturi
. Transizioni n * in composti carbonilici saturi.")
25
Cromoforo carbonilico In aldeidi e chetoni saturi le intense transizioni * sono localizzate nel lontano UV, mentre la transizione n *, di debole intensità, cade nellintervallo 275-230 nm. Gruppi auxocromi legati direttamente al carbonile spostano la transizione n * verso lunghezze donda più corte. Quando il groppo carbonilico è coniugato con un doppio legame la transizione * subisce un effetto batocromico più pronunciato rispetto a quella della transizione n *: negli enoni con il crescere della catena coniugata la transizione * alla massima lunghezza donda si sposta sempre più nettamente verso il visibile, raggiunge la n *e la copre. La posizione del picco è particolarmente influenzata dal solvente.

26
Cromoforo benzenico Il benzene presenta tre bande di assorbimento, di cui quella a lunghezza donda maggiore è di debole intensità (banda B) e in alcuni solventi assume una tipica struttura fine vibrazionale. Come per gli altri gruppi cromofori la sostituzione con gruppi auxocromi o cromofori determina una variazione dellintensità e posizione delle bande. In ogni caso si ha un effetto batocromico di tutte le bande, spesso associato con un effetto ipercromico. 180220240260 (nm)
.")
27
Cromoforo benzenico

28
APPLICAZIONI Analisi Quantitativa: (in assenza di scostamenti dalla legge di Lambert-Beer) se per una data sostanza è noto il coefficiente di estinzione molare la misura della assorbanza permette di ricavare direttamente la sua concentrazione Analisi Strutturale: può fornire informazioni sulla presenza o assenza nella molecola di alcuni gruppi cromofori
 se per una data sostanza è noto il coefficiente di estinzione molare la misura della assorbanza permette di ricavare direttamente la sua concentrazione Analisi Strutturale: può fornire informazioni sulla presenza o assenza nella molecola di alcuni gruppi cromofori")
29
APPLICAZIONI Analisi di miscele: (in assenza di scostamenti dalla legge di Lambert-Beer) la composizione di una miscela di due/tre componenti può essere ricavata se esse hanno massimi di assorbimento a lunghezze donda diverse. Ad esempio per una miscela di due sostanze, A e B, che hanno massimo di assorbimento rispettivamente in 1 e 2 si misura lassorbanza in corrispondenza di tali lunghezzo donda, e si ricavano le concentrazioni risolvendo il sistema di equazioni:

30
APPLICAZIONI Determinazione di costanti di equilibrio: il principio è lo stesso dellanalisi delle miscele. Ad esempio in un equilibrio acido-base del tipo: le due specie A - e AH presentano solitamente spettri UV diversi e le loro concentrazioni possono essere determinate in maniera analoga a quella di due componenti di una miscela. Se è noto il pH la K a può essere ricavata in maniera semplice.

31
APPLICAZIONI Determinazione di costanti di equilibrio (continua) Si noti che per essere certi che stiamo osservando un equilibrio tra due specie bisogna eseguire le misure in varie soluzioni a valori diversi di pH, ma con la stessa concentrazione totale di acido e verificare la presenza di un punto (punto isosbestico) in cui tutte le curve di assorbimento si intersecano
 Si noti che per essere certi che stiamo osservando un equilibrio tra due specie bisogna eseguire le misure in varie soluzioni a valori diversi di pH, ma con la stessa concentrazione totale di acido e verificare la presenza di un punto (punto isosbestico) in cui tutte le curve di assorbimento si intersecano")
32
APPLICAZIONI Studi cinetici La maggior parte degli spettrofotometri consente di registrare lassorbanza in funzione del tempo. Se in soluzione sta avvenendo una reazione chimica è possibile seguire le variazioni di concentrazione di una particolare sostanza (un reagente che sta scomparendo o un prodotto che si sta formando) e da questa ricavare la velocità con cui sta avvenendo la reazione data.
 e da questa ricavare la velocità con cui sta avvenendo la reazione data..")










 tra la radiazione.>")









