Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
TERMODINAMICA STATISTICA APPLICAZIONI

2
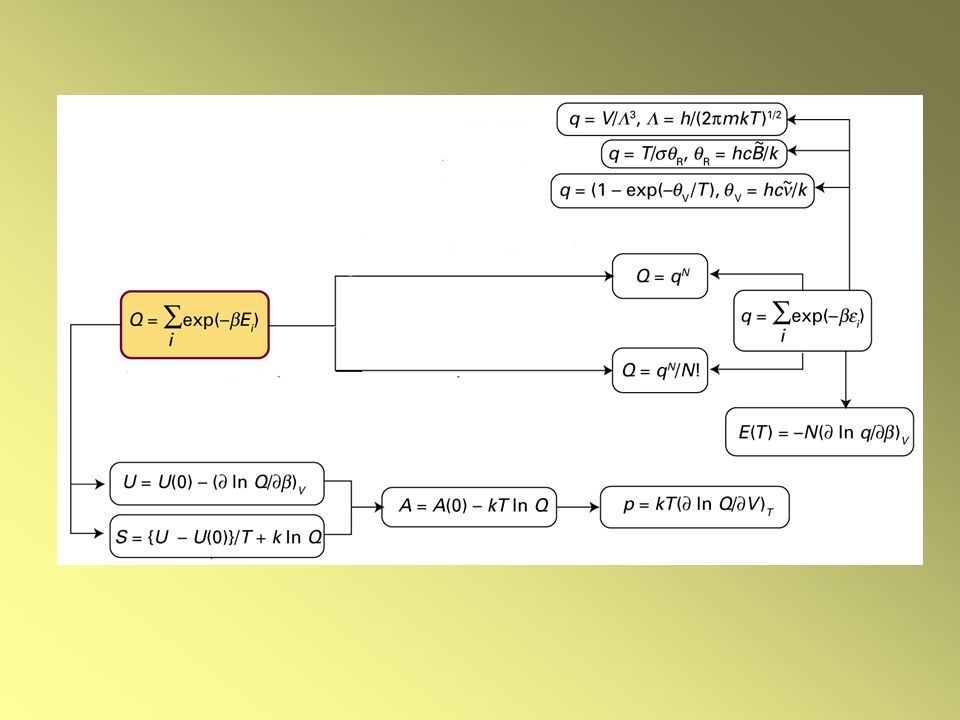
RELAZIONI FONDAMENTALI
LE FUNZIONI TERMODINAMICHE Energia interna Entropia Se le molecole sono indipendenti e distinguibili Q = qN indistinguibili Q = qN/N!

3
Capacità termica a volume costante cv

4
Energia di Helmholtz A = U – T S A(0) = U(0)
 = U(0)")
5
Pressione Entalpia A = U – T S dA = -p dV – S dT
a T costante p = - (A / V)T Equazione di stato Entalpia H = U + PV
T. Equazione di stato. Entalpia. H = U + PV.")
6
Energia di Gibbs G = H – T S = U + PV – T S = A + PV
Per un gas perfetto PV = RT G = A + RT

8
Funzione di partizione molecolare
L’energia totale tot di una molecola poliatomica può essere approssimata come somma di energia traslazionale ed energia interna: Emolecolare = Etraslazionale + Einterna L’energia interna si può approssimare come somma Einterna = Erotazionale + Evibrazionale + Eelettronica La separazione è esatta per il moto traslazionale. Il moto vibrazionale è indipendente dal: moto rotazionale nell’approssimazione rotore rigido. moto elettronico nell’approssimazione Born-Oppenheimer.

9
Separabilità delle energie
Se l’energia totale di una molecola i è

10
Fattorizzazione dei modi energetici
Supponiamo ci siano 2 soli modi e (rotazionale e vibrazionale per esempio) A ciascun modo sono associati stati energetici ed i corrispondenti numeri quantici - modo : numero quantico k - modo : numero quantico r
 A ciascun modo sono associati stati energetici ed i corrispondenti numeri quantici. - modo : numero quantico k. - modo : numero quantico r.")
11
La funzione di partizione qtot:

12
i termini in parentesi in ciascuna riga sono identici e formano la somma:

13
Il contributo traslazionale
L’espressione di qT è stata ricavata nell’approssimazione che gli stati formino un continuo ed è valida se la lunghezza d’onda termica Λ è piccola rispetto a V. H2 a 25 C Λ = nm O2 Λ = nm Per una molecola di O2 in un recipiente di 100 cm3 qT =

14
Il contributo rotazionale
Molecola biatomica AB non simmetrica (HCl) assunta come un rotore rigido. I momento di inerzia della molecola J numero quantico rotazionale 0,1,2,.. B costante rotazionale
 assunta come un rotore rigido. I momento di inerzia della molecola. J numero quantico rotazionale 0,1,2,.. B costante rotazionale.")
15
Temperatura rotazionale caratteristica r
Molecola r (K) H2 88 OH 27.5 HCl 9.4 CO 2.77 I2 0.053 J = 0 2kr 4kr J = 1 J = 2 Questa somma non può essere scritta in forma chiusa
 H OH HCl CO I J = 0. 2kr. 4kr. J = 1. J = 2. Questa somma non può essere scritta in forma chiusa.")
16
Calcolo esatto di qR T r
kT/hc = cm-1 a K 1H35Cl B= cm-1 hcB/kT = J 1 2.71 2 3.68 3 3.79 4 3.24 … 10 0.08 qR = 19.9 Sommando fino a J=50 qR =

17
Calcolo approssimato di qR T >> r
se nessun atomo nella molecola è troppo leggero (se il momento di inerzia non è troppo piccolo) se la temperatura non è troppo bassa (vicina a 0 K) T >> r gli stati rotazionali sono molto vicini
 se la temperatura non è troppo bassa (vicina a 0 K) T >> r gli stati rotazionali sono molto vicini.")
18
CO qrot somma qrot

19
questa equazione è corretta per le molecole biatomiche eteronucleari
per le molecole biatomiche omonucleari sovrastima di un fattore 2 gli stati rotazionali.

20
Quando una molecola lineare simmetrica ruota di 180o genera una configurazione indistinguibile da quella di partenza. Tutte le molecole biatomiche omonucleari Le molecole lineari simmetriche (CO2, C2H2) Numero di simmetria = 2 per le biatomiche omonucleari = 1 per le biatomiche eteronucleari = 2 per H2O, = 3 per NH3, = 12 per CH4 e C6H6
 Numero di simmetria = 2 per le biatomiche omonucleari. = 1 per le biatomiche eteronucleari. = 2 per H2O, = 3 per NH3, = 12 per CH4 e C6H6.")
21
Proprietà rotazionali di molecole a 300 K
qr (K) s T/qr qrot H CH HCl HI N CO CO I
 s T/qr qrot. H CH HCl HI N CO CO I")
22
Conclusioni I livelli energetici rotazionali, sebbene molto più spaziati dei livelli energetici traslazionali, sono ancora abbastanza vicini da permetterci in generale di usare l’approssimazione del continuo e di sostituire la sommatoria con un’integrazione. Nel calcolo della funzione di partizione rotazionale occorre tenere conto del numero di orientazioni equivalenti mediante il numero di simmetria.

23
Il contributo vibrazionale
Approssimazione dell’oscillatore armonico Se poniamo a zero l’energia dello stato fondamentale, ovvero misuriamo l’energia a partire dal livello fondamentale, le energie relative possono essere espresse come hv hv hv hv hv

24
Temperatura vibrazionale caratteristica v

25
Sostanza v (K) H2 6140 OH 5360 HCl 4300 CH 4100 CO 3120 NO 2740 O2 2260 Cl2 810 I2 309
 H OH 5360 HCl 4300 CH 4100 CO 3120 NO 2740 O Cl2 810 I2 309")
26
Dipendenza dalla Temperatura della funzione di partizione vibrazionale
Per T 0 qV = 1 Per T >> v andamento lineare

27
La spaziatura dei livelli rotazionali è più grande di ordini di grandezza di quella dei livelli traslazionali. La spaziatura dei livelli vibrazionali è più grande almeno di un ordine di grandezza di quella dei livelli rotazionali non si può semplificare usando l’approssimazione del continuo non si ha eccitazione apprezzabile a T ambiente a 300 K qV ≈ 1 per molecole leggere

28
Specie v (K) qV (300 K) H2 6140 1.000 HD 5226 D2 4307 N2 3352 CO 3120 Cl2 810 1.075 I2 309 1.556
 qV (300 K) H HD 5226 D N CO 3120 Cl I")
29
Il contributo elettronico
Le temperature elettroniche caratteristiche, el, sono dell’ordine di decine di migliaia di kelvin. Gli stati elettronici eccitati rimangono non popolati a meno che la temperatura raggiunga alcune migliaia di kelvin. Solo il primo termine della funzione di partizione elettronica deve essere preso in considerazione per temperature nell’intervallo T ambiente – T moderatamente alta. Posto 0E = 0

30
Metalli alcalini: stati di doppietto qE = 2
Eccezione: in alcuni casi occorre tener conto della degenerazione dello stato elettronico fondamentale Gli atomi hanno spesso stati degeneri (P, D, … doppietti, tripletti, …) Metalli alcalini: stati di doppietto qE = 2 Tra le molecole O2 è un esempio: ha uno stato fondamentale 3 volte degenere.
 Metalli alcalini: stati di doppietto qE = 2. Tra le molecole O2 è un esempio: ha uno stato fondamentale 3 volte degenere.")
31
Funzione di partizione elettronica
Specie Simbolo del termine gn Δel (K) Li 2S1/2 g0=2 C 3P0 g0=1 N 4S3/2 g0=4 O 3P2 g0=5 F 2P3/2 2P1/2 g1=2 590 NO 21/2 2 3/2 178 O2 3-g g0=3 1Δg g1=1 11650
 Li. 2S1/2. g0=2. C. 3P0. g0=1. N. 4S3/2. g0=4. O. 3P2. g0=5. F. 2P3/2. 2P1/2. g1= NO. 21/2. 2 3/ O2. 3-g. g0=3. 1Δg. g1=")
32
NO ….1 Momento angolare orbitale dell’elettrone: rotazione attorno all’asse di legame in senso orario ed in senso antiorario = 1 Λ = Spin ha 2 orientazioni rispetto all’asse di legame / /2 Accoppiamento spin-orbita Se i due momenti sono paralleli J = 3/2 23/2 Se antiparalleli J = 1/2 21/2 Separazione piccola: 4 stati accessibili

33
T = qE = 2 T elevata qE = 4 T = 298 K qE = 3.1

34
Contributi alla funzione di partizione molecolare
Molecola biatomica

36
Calcolo delle grandezze termodinamiche
Energie medie Capacità termiche Equazioni di stato

37
Energie medie Energia media traslazionale
Particella in una scatola di lunghezza X

38
Per una molecola in 3 dimensioni
Non dipende dalle dimensioni del contenitore. L’energia interna di un gas perfetto non dipende dal volume. In accordo con il principio di equipartizione classico il contributo all’energia media per ogni termine quadratico è ½ k T

39
Energia media rotazionale
Molecola biatomica A bassa T T < R occorre sommare termine a termine

40
Ad alta T T >> R Risultato in accordo con il teorema di equipartizione dell’energia. Ecin = ½ I a2 + ½ I b2 2 termini quadratici nell’espressione dell’energia. <E> = 2 (½ k T) = kT
 = kT.")
41
Energia media vibrazionale
Andamento a bassa T

42
Ad alta T T >> V E = ½ mvx2 + ½ k x2 2 termini quadratici <V> = 2 (½ k T) = kT Andamento lineare ad alta T
 = kT. Andamento lineare ad alta T.")
43
Capacità termiche Se l’energia interna è somma di contributi TOT T + R + V + E

44
Contributi alla capacità termica
Per un gas monoatomico cV = J K-1 mole-1 ad alta T A bassa T occorre utilizzare l’espressione esatta di <εR> e derivarla rispetto a T

45
Solo ad alta temperatura Uvib = RT cV = R
Formula di Einstein

46
Ad alte temperature Quando T ~ V cV/R ~ 1

47
Capacità termica totale
3 modi traslazionali attivi in un gas 3/2 R R* modi rotazionali attivi R*= 2 molecole lineari = 3 molecole non lineari ½ R* R V* modi vibrazionali attivi V* R

48
Ogni modo diventa attivo quando si supera la sua T caratteristica.
cV diventa molto grande alla dissociazione perché l’energia è utilizzata per causare la rottura del legame e non per aumentare T. Infine cV ritorna a 2 volte il valore traslazionale per i due atomi.

49
Capacità termica totale di H2O vapore a 100 C
I 3 gradi di libertà traslazionali danno un contributo 3/2 R = 12.5 J K-1 I 3 gradi di libertà rotazionali hanno temperature caratteristiche 40 K, 21 K e 13 K: sono eccitati ed il loro contributo è 3/2 R = 12.5 J K-1 Le temperature vibrazionali caratteristiche sono 5300 K, 2300 K e 5400 K: le vibrazioni non sono eccitate. La capacità termica totale è predetta 25 J K-1 Il valore sperimentale è 26.1 J K-1 La differenza è dovuta probabilmente alla deviazione da gas perfetto.

50
energia media capacità termica
Modo generale Traslazione Rotazione (T>>R) Vibrazione Vibrazione(T>>V)
 Vibrazione. Vibrazione(T>>V)")
51
Equazioni di stato Per un gas ideale Q = qN / N! con q = V/Λ3

52
N = n NA L’equazione di stato di un gas di particelle indipendenti è la legge dei gas perfetti

53
Per un gas reale Etot = Ecin + Epot Quindi la funzione di partizione è il prodotto di un termine legato all’energia cinetica ed un termine legato all’energia potenziale. L’energia cinetica è la somma delle energie cinetiche delle singole molecole. La funzione di partizione canonica Q è il prodotto di un termine identico a quello dei gas perfetti ed un termine, detto integrale configurazionale Z, che dipende dal potenziale intermolecolare.

54
Ogni configurazione, cioè ogni disposizione di molecole, ha una probabilità di realizzarsi data da una distribuzione di Boltzmann in cui l’esponente è dato dall’energia potenziale. Gas ideale Epot = 0 Z = VN/N! Ma per un insieme di particelle indipendenti dotate di solo moto traslazionale Q = qN / N! con q = qT = V/Λ3 Q = VN / Λ3N N! = Z / Λ3N

55
INTERAZIONI MOLECOLARI NEI LIQUIDI
Gas perfetti sistemi completamente disordinati Solidi cristallini sistemi completamente ordinati Liquidi ordine solo a corto raggio Funzione di distribuzione radiale g(r) Numero di atomi in una crosta sferica di spessore dr a distanza r da un atomo di riferimento 4 π r2 dr ρ g(r) dove ρ = N / V densità
 Numero di atomi in una crosta sferica di spessore dr a distanza r da un atomo di riferimento. 4 π r2 dr ρ g(r) dove ρ = N / V densità.")
56
GAS

57
SOLIDO

58
LIQUIDO

59
Funzione di distribuzione radiale g(r)
gO-O nell’acqua a 3 temperature

60
Calcoli di g(r) Funzione di distribuzione radiale g Densità alta bassa La struttura dei liquidi è dominata dalla parte repulsiva del potenziale, la parte attrattiva modifica questa struttura base talora molto debolmente.

61
Condizioni periodiche al contorno
Non potendo trattare numeri troppo grandi di molecole, per evitare gli effetti di bordo che sarebbero prevalenti con poche (103) molecole, si introducono repliche identiche in modo da riempire tutto lo spazio. Se una molecola esce da una faccia, un’altra entra dalla faccia opposta.
 molecole, si introducono repliche identiche in modo da riempire tutto lo spazio. Se una molecola esce da una faccia, un’altra entra dalla faccia opposta.")
62
Simulazione dei liquidi
Monte Carlo Configurazioni del liquido generate mediante l’uso di numeri casuali <A> = Ai pi Media pesata su tutti i membri dell’insieme Dinamica Molecolare Una configurazioni del liquido evolve nel tempo secondo l’equazione di Newton F = m a <A> Media sui valori incontrati ai vari tempi ti

63
Entropia residua L’entropia alla temperatura T può essere determinata con misure calorimetriche Il valore sperimentale dell’entropia potrebbe essere minore del valore calcolato a partire dalla funzione di partizione. Disordine è presente nel solido anche a T=0. L’entropia a T=0 è talora > 0 ed è chiamata entropia residua. Temperatura Entropia

64
Esempio: un cristallo composto di molecole AB con A molto simile a B (per esempio CO).
…AB AB AB AB… e …AB BA BA AB … hanno energie quasi uguali. Ogni molecola può assumere le 2 orientazioni AB e BA. N = n NAV molecole 2N disposizioni con la stessa energia S = k ln W = k ln 2N = kN ln 2 = n R ln 2 R ln 2 = 5.8 J K-1 mol-1 CO entropia residua = 5 J K-1 mol-1

65
FClO3 F può puntare ai 4 vertici di un tetraedro
R ln 4 = J K-1 mol-1 entropia residua = J K-1 mol-1

66
H2O N molecole di H2O 2N atomi H.
Per ogni legame ad idrogeno l’atomo H ha 2 possibilità: O-H…O, O…H-O. 22N disposizioni complessive. Per l’atomo al centro in teoria ci sarebbero 24 = 16 possibilità.

67
In realtà la molecola centrale di riferimento, formato con un’altra molecola o un legame O-H…O oppure un legame O…H-O, può formare solo un secondo legame O-H con 3 altre molecole. 2 x 3 = 6 possibilità le disposizioni complessive sono 22N (6/16)N = (6/4)N = (3/2)N
N = (6/4)N = (3/2)N.")
68
Costanti di equilibrio
G = H – T S = U + pV – T S = A + pV Per un gas perfetto PV = nRT G = A + nRT Q = qN / N! N = n NAV funzione di partizione molare

69
Data la reazione a A + b B = c C + d D
Scelta po = 1 bar definiamo l’energia libera molare standard della specie J è la funzione di partizione molare standard della specie J Data la reazione a A + b B = c C + d D

70
Energia interna di reazione a T = 0 K
Poiché G(0) = U(0) Energia interna di reazione a T = 0 K D0 (reagenti) D0 (prodotti) Può essere calcolata conoscendo le energie di dissociazione di reagenti e prodotti
 = U(0) Energia interna di reazione a T = 0 K. D0 (reagenti) D0 (prodotti) Può essere calcolata conoscendo le energie di dissociazione di reagenti e prodotti.")
72
Reazione R → P L’occupazione dei livelli è data dalla distribuzione di Boltzmann indipendentemente dal sistema R o P a cui i livelli appartengono. La costante di equilibrio è la somma delle probabilità che il sistema si trovi in uno degli stati di P divisa per la somma delle probabilità che sia in uno degli stati di R

73
ΔE0 : differenza fra il più basso livello di R e di P

74
All’equilibrio tutti i livelli sono accessibili (dipendenza da T).
La composizione all’equilibrio dipende dalla distribuzione di Boltzmann complessiva. Se le spaziature dei livelli di R e P sono circa uguali, al crescere di ΔE0 la specie con livelli più bassi diventa dominante. Effetto del ΔHreazione

75
Anche se i livelli energetici di P sono più alti di quelli di R (ΔE0 grande), P può avere una densità di stati elevata la sua popolazione domina la miscela. La funzione di partizione qP >> qR domina K Effetto del ΔSreazione Entropia tanto importante quanto entalpia nel definire l’equilibrio.

76
Reazione endotermica R ha un solo livello qR = 1 P ha livelli equispaziati ΔE0 positivo e molto grande, esponenziale molto piccolo K << 1 R domina ΔE0 positivo e piccolo a basse T K < 1 ad alte T il termine kT/ può rendere K > 1

77
Na2 → 2 Na ΔEO = 0.73 eV exp (-ΔEO/kT) = 2.09 10-4 a 1000 K
Reazione endotermica ΔEO = 0.73 eV exp (-ΔEO/kT) = a 1000 K Dal punto di vista dell’energia l’equilibrio dovrebbe essere spostato verso i reagenti. K = a 1000 K Lo stato fondamentale di Na2 è un singoletto, mentre lo stato fondamentale di Na è un doppietto. Na ha due stati quasi degeneri ed un peso statistico molto più alto. Distribuzione di Boltzmann Popolazione Energia
 = a 1000 K. Dal punto di vista dell’energia l’equilibrio dovrebbe essere spostato verso i reagenti. K = a 1000 K. Lo stato fondamentale di Na2 è un singoletto, mentre lo stato fondamentale di Na è un doppietto. Na ha due stati quasi degeneri ed un peso statistico molto più alto. Distribuzione. di Boltzmann. Popolazione. Energia.")
Presentazioni simili






 Neutroni (n°) Elettroni (e) Gli atomi contengono diversi tipi di particelle subatomiche.>")


 Acidi poliprotici>")











