Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Farmacovigilanza

2
Non tutti i rischi possono essere individuati prima che il farmaco venga messo in commercio.
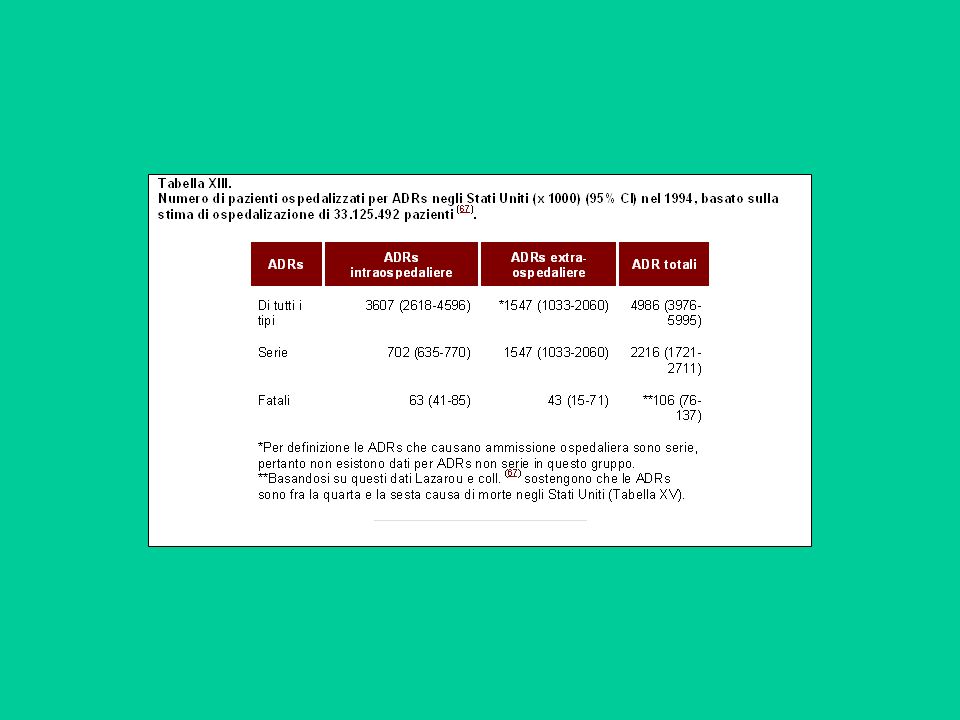
Né gli studi sugli animali né i trials clinici in pazienti riveleranno sempre tutti i possibili effetti collaterali di un farmaco. Questi potranno essere noti solo quando il farmaco sarà stato somministrato ad un gran numero di pazienti e per un prolungato periodo di tempo

3
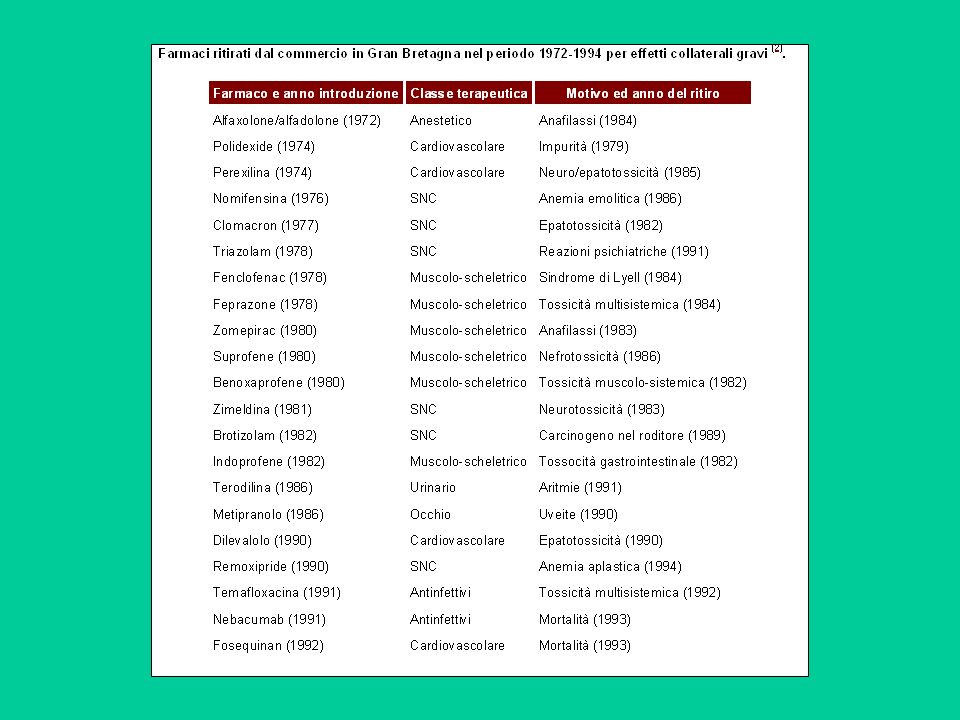
Il 51% dei farmaci approvati presentano gravi reazioni avverse non scoperte prima della approvazione alla commercializzazione.

5
ADRs: Adverse Drug Reactions

7
Gli studi animali hanno molte limitazioni nella loro capacità di predire la tossicità umana
Esempio: practololo (ß1-bloccante) per alcuni anni ampiamente impiegato in terapia. Il farmaco nell'uomo ha causato una sindrome definita "oculomucosocutanea" caratterizzata da dermatite, cheratocongiuntivite e peritonite sclerosante, che ha portato nel 1976 al ritiro del farmaco dal commercio. Questa sindrome non era mai stata osservata negli studi pre-clinici: successive indagini condotte su animali di piccola taglia (sia quelli che metabolizzano il practololo in maniera simile all'uomo, sia quelli che lo metabolizzano in maniera molto più estesa) hanno dimostrato che non esiste un modello animale in cui sia possibile osservare questa reazione
 per alcuni anni ampiamente impiegato in terapia. Il farmaco nell uomo ha causato una sindrome definita oculomucosocutanea caratterizzata da dermatite, cheratocongiuntivite e peritonite sclerosante, che ha portato nel 1976 al ritiro del farmaco dal commercio. Questa sindrome non era mai stata osservata negli studi pre-clinici: successive indagini condotte su animali di piccola taglia (sia quelli che metabolizzano il practololo in maniera simile all uomo, sia quelli che lo metabolizzano in maniera molto più estesa) hanno dimostrato che non esiste un modello animale in cui sia possibile osservare questa reazione.")
8
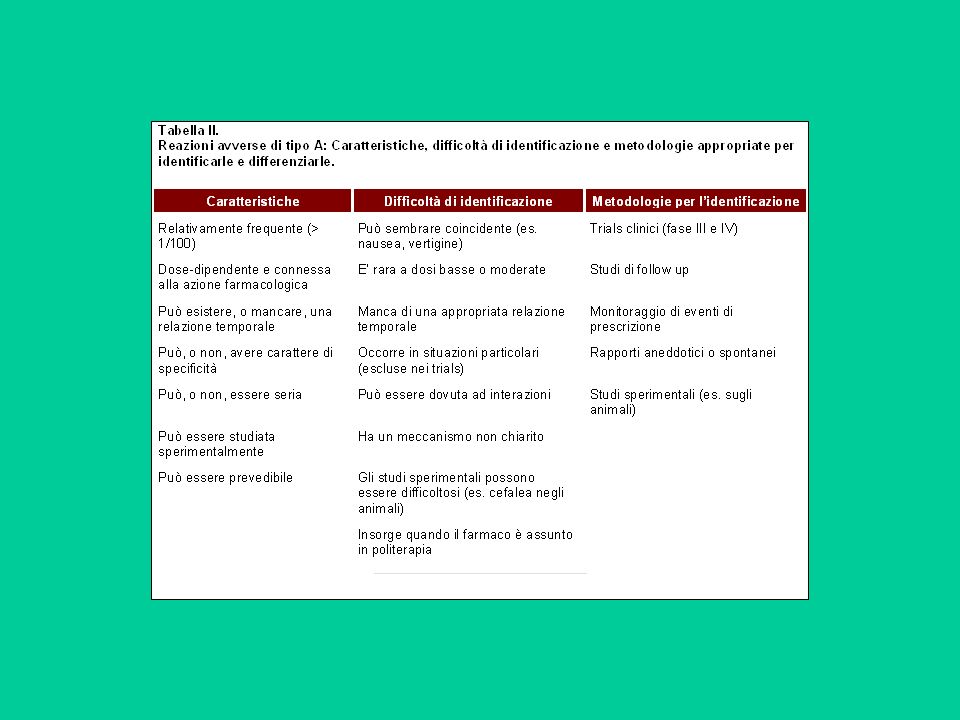
I trials clinici non sono in grado di rivelare tutte le reazioni avverse al farmaco perché:
numero ristretto di pazienti. Il numero di soggetti su cui viene studiato un farmaco, fra volontari sani e volontari pazienti, non supera le unità. Per avere il 95% di probabilità di individuare una o più reazioni avverse, in base all'incidenza della stessa reazione avversa sono necessari spesso numeri molto elevati. Ne consegue che solo reazioni avverse con incidenza approssimativa di 1 a 1000 o maggiori possono essere individuate negli studi pre-marketing.

10
breve durata della sperimentazione clinica
breve durata della sperimentazione clinica. Per farmaci che saranno di uso cronico (es. antiepilettici, antiinfiammatori, antiipertensivi, ecc.) la sperimentazione pre-marketing non potrà mai essere condotta per il periodo di tempo (anni, tutta la vita del paziente, ecc.) in cui è prevedibile che il farmaco verrà poi utilizzato. popolazione selezionata. Nell'interesse del paziente volontario, e dello sperimentatore, gli studi clinici sono condotti selezionando i pazienti sulla base di criteri di inclusione ed esclusione. Verranno pertanto volontariamente esclusi neonati, donne in gravidanza, pazienti molto anziani o con patologie complicanti o con politerapie (es. pazienti con trapianto o con ipercolesterolemia, come nel su citato caso del mibefradil), ecc.
 la sperimentazione pre-marketing non potrà mai essere condotta per il periodo di tempo (anni, tutta la vita del paziente, ecc.) in cui è prevedibile che il farmaco verrà poi utilizzato. popolazione selezionata. Nell interesse del paziente volontario, e dello sperimentatore, gli studi clinici sono condotti selezionando i pazienti sulla base di criteri di inclusione ed esclusione. Verranno pertanto volontariamente esclusi neonati, donne in gravidanza, pazienti molto anziani o con patologie complicanti o con politerapie (es. pazienti con trapianto o con ipercolesterolemia, come nel su citato caso del mibefradil), ecc.")
11
indicazione ristretta
indicazione ristretta. Il farmaco verrà sperimentato nell'uomo in base alla indicazione per cui è stato previsto e precedentemente studiato negli animali. L'amantadina (farmaco oggi utilizzato anche nel morbo di Parkinson) fu pensato come farmaco antivirale e gli studi pre-clinici furono ovviamente condotti allo scopo di dimostrare questa attività. Pertanto in tali studi furono volutamente esclusi i pazienti con morbo di Parkinson. ambiente della sperimentazione. La maggior parte degli studi clinici viene condotta in strutture ospedaliere, spesso molto prestigiose, dove i pazienti sono sottoposti a regimi dietetici e comportamentali strettamente controllati ed il paziente non è libero di gestire le proprie abitudini (alimentari, voluttuarie, ecc.) secondo una propria scelta.
 fu pensato come farmaco antivirale e gli studi pre-clinici furono ovviamente condotti allo scopo di dimostrare questa attività. Pertanto in tali studi furono volutamente esclusi i pazienti con morbo di Parkinson. ambiente della sperimentazione. La maggior parte degli studi clinici viene condotta in strutture ospedaliere, spesso molto prestigiose, dove i pazienti sono sottoposti a regimi dietetici e comportamentali strettamente controllati ed il paziente non è libero di gestire le proprie abitudini (alimentari, voluttuarie, ecc.) secondo una propria scelta.")
12
Differenza tra studi clinici controllati (RCTs) e normale pratica clinica
breve termine gruppi a rischio esclusi ben definito precise e limitate 1 o pochi costante continuo precisa e rigorosa eventi ben raccolti numero di pazienti durata popolazione problema clinico indicazioni numero di farmaci dose profilo d’uso assistenza paziente follow-up fino a 106 lunga durata potenzialmente la popolazione generale spesso poco definito allargate a volte molti spesso variabile intermittente variabile meno accurato

13
J.P. Ioannidis, J.L. Lau JAMA 24/31, 2001
Negli studi clinici la sicurezza (safety) dei farmaci non è spesso sufficientemente controllata e riportata. Risultati dall’analisi di 192 trials Clinical adverse events Adequate reporting Partially adequate reporting Inadequate reporting Laboratory-defined toxicity Discontinuations due to toxicity Number per arm not given Reasons per arm not given No. (%) 75 (39) 22 (11) 95 (50) 56 (29) 15 (8) 121 (63) 49 (25) 104 (54) J.P. Ioannidis, J.L. Lau JAMA 24/31, 2001
 dei farmaci non è spesso sufficientemente controllata e riportata. Risultati dall’analisi di 192 trials. Clinical adverse events. Adequate reporting. Partially adequate reporting. Inadequate reporting. Laboratory-defined toxicity. Discontinuations due to toxicity. Number per arm not given. Reasons per arm not given. No. (%) 75 (39) 22 (11) 95 (50) 56 (29) 15 (8) 121 (63) 49 (25) 104 (54) J.P. Ioannidis, J.L. Lau JAMA 24/31,")
14
“The absolute space allocated to safety information was limited (median 0.3 page, interquartile range page). Overall the space given to safety information was the same as or less than the space given for the names of Authors and their affiliations”

16
La farmacovigilanza ha 4 obiettivi principali:
riconoscere, il più rapidamente possibile, nuove ADRs migliorare ed allargare le informazioni su ADRs sospette o già note valutare i vantaggi di un farmaco su altri o su altri tipi di terapia comunicare l'informazione in modo da migliorare la pratica terapeutica.

17
Decisioni normative (ritiro, limitazioni e precauzioni d’uso)
Il principale obiettivo della farmacovigilanza, riconoscendo il più precocemente possibile nuove ADRs, è quello di fornire un precoce segnale d’allarme : a) generazione di una ipotesi; b) rafforzamento dell'ipotesi e valutazione preliminare dei dati disponibili; c) verifica, valutazione e spiegazione del segnale Decisioni normative (ritiro, limitazioni e precauzioni d’uso)
 generazione di una ipotesi; b) rafforzamento dell ipotesi e valutazione preliminare dei dati disponibili; c) verifica, valutazione e spiegazione del segnale. Decisioni normative (ritiro, limitazioni e precauzioni d’uso)")
18
Ciclo di vita di un medicinale
Preclinica Clinical trials Immissione in commercio Sicurezza clinica Sorveglianza Pratica clinica Passiva Farmacovigilanza Identificazione Segnali di allarme Strumenti: Segnalazioni spontanee Letteratura Valutazione di serie di casi Attiva Farmacoepidemiologia Verifica/conferma Quantificazione dei rischi Strumenti: Studi descrittivi Studi analitici

19
Letteratura scientifica
PSUR Studi ad hoc Segnalazioni spontanee GESTIONE ORGANIZZAZIONE INTEGRAZIONE Sistemi di FV altri Paesi Uso nella popolazione Problematiche emergenti VALUTAZIONE INTERVENTI REGOLATORI COMUNICAZIONE Autorità europee CUF

22
CRITICHE ALLA DEFINIZIONE “CLASSICA” DI REAZIONE AVVERSA
il termine “dannosa” è troppo vago, e rischia di includere anche reazioni decisamente minori. le ADRs dovute ad “errori” di trattamento non sono espressamente considerate le potenziali reazioni avverse da contaminanti (ad es. in preparazioni erboristiche) o da eccipienti non sono incluse nella dizione “ADRs”
 o da eccipienti non sono incluse nella dizione ADRs")
23
PROPOSTA DI EDWARDS e ARONSON (2000)
“An appreciably harmful or unpleasant reaction, resulting from an intervention related to the use of a medicinal product, which predicts hazard from future administration and warrants prevention or specific treatment, or alteration of dosage regimen, or withdrawl of the product”.

32
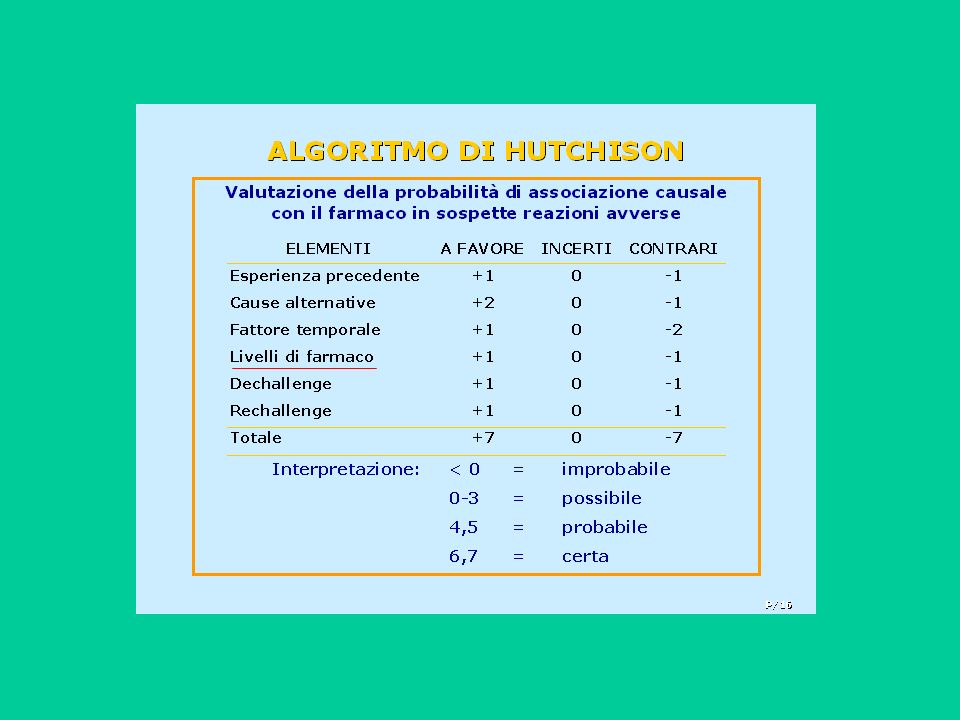
Come stabilire se l’evento osservato è stato causato effettivamente dal farmaco?

33
I procedimenti utilizzabili per stabilire il nesso di causalità tra farmaco ed evento sono diversi:
- giudizio clinico - utilizzo di linee-guida per patologia - applicazione di una serie di criteri di tipo “farmacologico” - utilizzo di alberi decisionali o di algoritmi formali

37
Qualunque metodo si scelga, gli elementi fondamentali da prendere in considerazione sono i seguenti:
- intervallo temporale - presenza di cause alternative - risposta alla sospensione (dechallenge) - risposta alla risomministrazione (rechallenge)
 - risposta alla risomministrazione (rechallenge)")
39
GENERAZIONE DI UN SEGNALE APPROFONDIMENTO FARMACOLOGICO
REAZIONE AVVERSA GENERAZIONE DI UN SEGNALE APPROFONDIMENTO FARMACOLOGICO STUDI EPIDEMIOLOGICI CONFERMA DELL’IPOTESI IDENTIFICAZIONE DI EVENTUALI FATTORI DI RISCHIO

40
Decisioni normative (ritiro, limitazioni e precauzioni d’uso)
segnale d’allarme : generazione di una ipotesi “il farmaco è legato ad un determinato effetto avverso” rafforzamento dell'ipotesi (altre segnalazioni, altre informazioni sul farmaco) e valutazione preliminare dei dati disponibili verifica e valutazione (e spiegazione) del segnale Decisioni normative (ritiro, limitazioni e precauzioni d’uso)
 e valutazione preliminare dei dati disponibili verifica e valutazione (e spiegazione) del segnale. Decisioni normative (ritiro, limitazioni e precauzioni d’uso)")
41
VALUTAZIONE DELLA POSSIBILITÀ DI CONTROLLARE IL RISCHIO
RIVALUTAZIONE DEL RAPPORTO RISCHIO/BENEFICIO (ANCHE IN CONSIDERAZIONE DI POSSIBILI ALTERNATIVE TERAPEUTICHE) FAVOREVOLE SFAVOREVOLE DEAR DOCTOR LETTER MODIFICA POSOLOGIA E/O INDICAZIONI RITIRO CONTROINDICAZIONI AVVERTENZE SPECIALI STRETTO MONITORAGGIO
 FAVOREVOLE SFAVOREVOLE. DEAR DOCTOR LETTER MODIFICA POSOLOGIA. E/O INDICAZIONI RITIRO. CONTROINDICAZIONI. AVVERTENZE SPECIALI. STRETTO MONITORAGGIO.")
42
Diagramma di flusso delle segnalazioni spontanee
1 segnalato a 1 sospetto a riconosciuto non segnalato b a 1 Evento non attribuito a 2 non riconosciuto b RICONOSCIMENTO ATTRIBUZIONE SEGNALAZIONE a + b = incidenza vera a 1 = a - 2 b - = reports spontanei

43
Le Segnalazioni Spontanee
Fattori che influenzano il reporting, a parità di altre condizioni (atteggiamento/sensibilità dei medici, organizzazione del sistema, ecc. ecc.): Elementi necessari per una valutazione: la rilevanza e la gravità degli eventi la novità dell’evento come ADR la novità del farmaco il rischio (incidenza) di base dell’evento nella popolazione il numero di pazienti trattati in termini di mesi/persona l numero di casi segnalati in un intervallo temporale
: Elementi necessari per una valutazione: la rilevanza e la gravità degli eventi. la novità dell’evento come ADR. la novità del farmaco. il rischio (incidenza) di base dell’evento nella popolazione. il numero di pazienti trattati in termini di mesi/persona. l numero di casi segnalati in un intervallo temporale.")
44
Quale livello ottimale di segnalazioni?
300 segnalazioni anno/milione di abitanti; 30% di natura grave e provenienti da almeno il 10% dei medici. In queste condizioni la comunità può essere ragionevolmente sicura che ADR importanti siano identificate in un tempo ragionevolmente breve. In Italia 120 segnalazioni anno/milione di abitanti

45
Decreto Legislativo 8 aprile 2003, n. 95
"Attuazione della direttiva 2000/38/CE relativa alle specialità medicinali" Art. 1. Art Il sistema nazionale di farmacovigilanza fa capo alla Direzione generale per la valutazione dei medicinali e per la farmacovigilanza del Ministero della salute, di seguito denominato "Direzione". 2. La Direzione…: a) raccoglie e valuta informazioni utili per la sorveglianza dei medicinali con particolare riguardo alle reazioni avverse, all'uso improprio, nonche' all'abuso degli stessi tenendo conto anche dei dati relativi ai consumi dei medesimi; b) promuove il processo di informatizzazione di tutti i flussi di dati necessari alla farmacovigilanza gestendo e coordinando, in particolare, la rete telematica nazionale di farmacovigilanza, che collega le strutture sanitarie, le regioni e le aziende farmaceutiche ecc.
 raccoglie e valuta informazioni utili per la sorveglianza dei medicinali con particolare riguardo alle reazioni avverse, all uso improprio, nonche all abuso degli stessi tenendo conto anche dei dati relativi ai consumi dei medesimi; b) promuove il processo di informatizzazione di tutti i flussi di dati necessari alla farmacovigilanza gestendo e coordinando, in particolare, la rete telematica nazionale di farmacovigilanza, che collega le strutture sanitarie, le regioni e le aziende farmaceutiche. ecc.")
46
3. Le regioni, singolarmente o di intesa fra loro, collaborano con la Direzione nell'attivita' di farmacovigilanza, fornendo elementi di conoscenza e valutazione ad integrazione dei dati che pervengono alla Direzione ai sensi dell'articolo 4. Le regioni, inoltre provvedono, nell'ambito delle proprie competenze, alla diffusione delle informazioni al personale sanitario ed alla formazione degli operatori nel campo della farmacovigilanza. Le regioni collaborano inoltre a fornire i dati sui consumi dei medicinali mediante programmi di monitoraggio sulle prescrizioni dei farmaci a livello regionale. Le regioni si possono avvalere per la loro attivita' anche di appositi Centri di farmacovigilanza.

47
«Art Il titolare dell'autorizzazione all'immissione in commercio e' tenuto a registrare in modo dettagliato tutte le sospette reazioni avverse da farmaci osservate in Italia, nell'Unione europea o in un Paese terzo. Il titolare dell'autorizzazione all'immissione in commercio e' tenuto, altresi', a registrare e a notificare immediatamente, e comunque entro quindici giorni solari da quando ne ha avuto notizia, qualunque sospetta reazione avversa grave da farmaci verificatasi in Italia segnalatagli da personale sanitario alla struttura sanitaria di appartenenza del segnalatore e, ove non fosse possibile identificare tale struttura, alla Direzione. Il titolare dell'autorizzazione all'immissione in commercio e' tenuto, altresi', a registrare e a notificare immediatamente, e comunque entro quindici giorni solari da quando ne ha avuto notizia, qualunque altra sospetta reazione avversa grave da farmaci di cui sia venuto a conoscenza alla Direzione. Eventuali aggiornamenti sulle segnalazioni di reazioni avverse ricevute possono essere richieste esclusivamente al Responsabile di farmacovigilanza mittente della segnalazione stessa quale il Ministero o la struttura sanitaria.

48
2. Il titolare dell'autorizzazione all'immissione in commercio provvede a che tutte le sospette reazioni avverse gravi ed inattese verificatesi nel territorio di un Paese terzo e segnalate da personale sanitario siano immediatamente e comunque entro quindici giorni solari da quando ne ha avuto notizia, notificate alla Direzione secondo le modalita' previste dal comma 5, lettera d). 3. Per i medicinali disciplinati dall'articolo 9, comma 3, del decreto legislativo 29 maggio 1991, n. 178, e dall'articolo 9-bis del decreto legislativo 18 febbraio 1997, n. 44, ai quali sono state applicate le procedure di mutuo riconoscimento e per i quali l'Italia e' il Paese membro di riferimento, il titolare dell'autorizzazione all'immissione in commercio provvede inoltre a segnalare alla Direzione, secondo le modalita' ed i tempi stabiliti in accordo con essa, qualunque sospetta reazione avversa grave verificatasi nella Comunita' europea.
. 3. Per i medicinali disciplinati dall articolo 9, comma 3, del decreto legislativo 29 maggio 1991, n. 178, e dall articolo 9-bis del decreto legislativo 18 febbraio 1997, n. 44, ai quali sono state applicate le procedure di mutuo riconoscimento e per i quali l Italia e il Paese membro di riferimento, il titolare dell autorizzazione all immissione in commercio provvede inoltre a segnalare alla Direzione, secondo le modalita ed i tempi stabiliti in accordo con essa, qualunque sospetta reazione avversa grave verificatasi nella Comunita europea..")
49
4. Il titolare dell'autorizzazione all'immissione in commercio di medicinali deve disporre, a titolo stabile e continuativo, di un responsabile del servizio di farmacovigilanza, in possesso della laurea in medicina e chirurgia o in farmacia, o in chimica e tecnologia farmaceutica, o in biologia o in chimica …. Le competenze del responsabile si estendono a tutti i medicinali della cui autorizzazione all'immissione in commercio e' titolare l'azienda da cui egli dipende, anche se commercializzati da altre aziende.

50
5. Il responsabile del servizio di farmacovigilanza assicura: a) l'istituzione e il funzionamento di un sistema atto a garantire che le informazioni su tutte le presunte reazioni avverse comunicate al personale della società ed agli informatori medico-scientifici, siano raccolte, ordinate e accessibili in un unico luogo; b) che tutte le informazioni relative alla sicurezza dei prodotti, successive all'atto dell'autorizzazione siano portate rapidamente a conoscenza del personale sanitario anche tramite i contatti del servizio di informazione scientifica della propria azienda; c) l'elaborazione dei rapporti di cui al successivo comma 6 [PSUR], da sottoporre alle autorità competenti .. d) la trasmissione.. per via telematica al sistema nazionale di farmacovigilanza, delle segnalazioni di sospette reazioni avverse gravi e inattese verificatesi in un Paese terzo..; e) la trasmissione alla struttura sanitaria di pertinenza delle segnalazioni di sospette reazioni avverse gravi o inattese avvenute sul territorio nazionale ricevute direttamente dal segnalatore e non tramite la rete nazionale di farmacovigilanza; f) la trasmissione, in maniera rapida ed esauriente, ad ogni richiesta della Direzione, di informazioni supplementari ai fini della valutazione dei rischi di un medicinale, comprese le informazioni riguardanti i volumi di vendita dello stesso.
![5. Il responsabile del servizio di farmacovigilanza assicura: a) l istituzione e il funzionamento di un sistema atto a garantire che le informazioni su tutte le presunte reazioni avverse comunicate al personale della società ed agli informatori medico-scientifici, siano raccolte, ordinate e accessibili in un unico luogo; b) che tutte le informazioni relative alla sicurezza dei prodotti, successive all atto dell autorizzazione siano portate rapidamente a conoscenza del personale sanitario anche tramite i contatti del servizio di informazione scientifica della propria azienda; c) l elaborazione dei rapporti di cui al successivo comma 6 [PSUR], da sottoporre alle autorità competenti ..](http://slideplayer.it/slide/593743/2/images/50/5.+Il+responsabile+del+servizio+di+farmacovigilanza+assicura%3A+a%29+l+istituzione+e+il+funzionamento+di+un+sistema+atto+a+garantire+che+le+informazioni+su+tutte+le+presunte+reazioni+avverse+comunicate+al+personale+della+societ%C3%A0+ed+agli+informatori+medico-scientifici%2C+siano+raccolte%2C+ordinate+e+accessibili+in+un+unico+luogo%3B+b%29+che+tutte+le+informazioni+relative+alla+sicurezza+dei+prodotti%2C+successive+all+atto+dell+autorizzazione+siano+portate+rapidamente+a+conoscenza+del+personale+sanitario+anche+tramite+i+contatti+del+servizio+di+informazione+scientifica+della+propria+azienda%3B+c%29+l+elaborazione+dei+rapporti+di+cui+al+successivo+comma+6+%5BPSUR%5D%2C+da+sottoporre+alle+autorit%C3%A0+competenti+...jpg "d) la trasmissione.. per via telematica al sistema nazionale di farmacovigilanza, delle segnalazioni di sospette reazioni avverse gravi e inattese verificatesi in un Paese terzo..; e) la trasmissione alla struttura sanitaria di pertinenza delle segnalazioni di sospette reazioni avverse gravi o inattese avvenute sul territorio nazionale ricevute direttamente dal segnalatore e non tramite la rete nazionale di farmacovigilanza; f) la trasmissione, in maniera rapida ed esauriente, ad ogni richiesta della Direzione, di informazioni supplementari ai fini della valutazione dei rischi di un medicinale, comprese le informazioni riguardanti i volumi di vendita dello stesso.")
51
6. …E' fatto obbligo al titolare dell'autorizzazione all'immissione in commercio di presentare alle autorità competenti le informazioni sulle sospette reazioni avverse in forma di rapporti periodici di aggiornamento sulla sicurezza o immediatamente su richiesta, oppure ad intervalli regolari come da schema seguente: ogni sei mesi per i primi due anni dalla data di rilascio della prima autorizzazione internazionale e successivamente ogni anno per i successivi due anni e in coincidenza del primo rinnovo dell'autorizzazione. In seguito tali rapporti periodici devono essere presentati ogni cinque anni congiuntamente alla domanda di rinnovo dell'autorizzazione. Essi devono contenere una valutazione scientifica dei benefici e dei rischi del medicinale in questione….. I rapporti periodici di aggiornamento sulla sicurezza - PSUR - sono presentati secondo la scadenza prevista, sulla base delle modalita' stabilite dalla Direzione.

52
7. Le Aziende titolari di autorizzazioni all'immissione in commercio comunicano al Ministero della salute, ufficio di farmacovigilanza, qualsiasi iniziativa adottata da altri competenti organismi sui propri prodotti per motivi di sicurezza, prima che tali interventi siano resi di dominio pubblico. 8. E' fatto obbligo al titolare dell'autorizzazione all'immissione in commercio di diffondere ai medici prescrittori le note informative e gli aggiornamenti sulla sicurezza dei farmaci, secondo indicazioni, tempi e modalità stabilite dalla Direzione, ogni qualvolta emergano nuove informazioni relative al profilo di tollerabilità del prodotto. 9. Le aziende titolari di autorizzazioni all'immissione in commercio di farmaci sono tenute a trasmettere trimestralmente per via informatica i dati di vendita delle specialita' medicinali utilizzando la procedura prevista dal decreto dirigenziale in data 24 maggio 2002 pubblicato nella Gazzetta Ufficiale n. 132 del 7 giugno 2002.

53
Art Le strutture sanitarie - Aziende unita' sanitarie locali, Aziende ospedaliere, Istituti di ricovero e cura a carattere scientifico - devono nominare un responsabile di farmacovigilanza della struttura che provvede a registrarsi alla rete nazionale di farmacovigilanza al fine dell'abilitazione necessaria per la gestione delle segnalazioni. Le strutture sanitarie private, al fine di assolvere ai compiti di farmacovigilanza, fanno riferimento al responsabile di farmacovigilanza della Azienda unita' sanitaria locale competente per territorio. 2. I medici e gli altri operatori sanitari sono tenuti a segnalare tutte le sospette reazioni avverse gravi o inattese di cui vengano a conoscenza nell'ambito della propria attivita'. Vanno comunque segnalate tutte le sospette reazioni avverse osservate, gravi, non gravi, attese ed inattese da tutti i vaccini e da farmaci posti sotto monitoraggio intensivo ed inclusi in elenchi pubblicati periodicamente dal Ministero della salute.

54
Per ulteriori informazioni sulla farmacovigilanza:

Presentazioni simili




























 Qualità. I progetti selezionati per l'erogazione di un finanziamento devono dimostrarsi di elevato.>")



