Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
Allineamento di sequenze Perché è importante? Le caratteristiche funzionali delle molecole biologiche dipendono dalle conformazione tridimensionale che gli atomi costituenti assumono nello spazio. Questa a sua volta dipende dalla sequenza delle unità elementari (in genere si tratta di aminoacidi, ma lo stesso discorso vale anche per i nucleotidi). Molecole che hanno sequenza primaria simile tendono ad avere strutture secondarie e terziarie simili. Se due proteine sono identiche al 50%, è altamente probabile che la loro struttura tridimensionale sia quasi completamente sovrapponibile.
. Molecole che hanno sequenza primaria simile tendono ad avere strutture secondarie e terziarie simili. Se due proteine sono identiche al 50%, è altamente probabile che la loro struttura tridimensionale sia quasi completamente sovrapponibile..")
2
Sequenze identiche al 62% 10 20 30 40 50 60 Chymotrypsin MLGITVLAALLACASSCGVPSFPPNLSARVVGGEDARPHSWPWQISLQYLKNDTWRHTCG :..:..:.: : ::: :..::.. ::::::.:::.:::::.::::.: : :::: Elastase MIRTLLLSTLVAGALSCGDPTYPPYVT-RVVGGEEARPNSWPWQVSLQYSSNGKWYHTCG 10 20 30 40 50 70 80 90 100 110 Chymotrypsin GTLIASNFVLTAAHCISNTRTYRVAVGKNNLEVEDEEGSLFVGVDTIHVHKRWNALLLR- :.:::...:::::::::..:::::..:..:: : : ::: :.:. : ::: ::.. Elastase GSLIANSWVLTAAHCISSSRTYRVGLGRHNLYVA-ESGSLAVSVSKIVVHKDWNSNQISK 60 70 80 90 100 110 120 130 140 150 160 170 Chymotrypsin -NDIALIKLAEHVELSDTIQVACLPEKDSLLPKDYPCYVTGWGRLWTNGPIADKLQQGLQ :::::.:::. : :.: ::.::::..::..::::::::::: :::. : :::: Elastase GNDIALLKLANPVSLTDKIQLACLPPAGTILPNNYPCYVTGWGRLQTNGAVPDVLQQGRL 120 130 140 150 160 170 180 190 200 210 220 230 Chymotrypsin PVVDHATCSRIDWWGFRVKKTMVCAGGDGVISACNGDSGGPLNCQLENGSWEVFGIVSFG :::.:::: ::: ::.:.:::::::::.::::::::::::.: :.: :::::: Elastase LVVDYATCSSSAWWGSSVKTSMICAGGDGVISSCNGDSGGPLNCQASDGRWQVHGIVSFG 180 190 200 210 220 230 240 250 260 Chymotrypsin SRRGCNTRKKPVVYTRVSAYIDWINEKM-QL :: :::.:: :.:::: ::::::.. Elastase SRLGCNYYHKPSVFTRVSNYIDWINSVIANN 240 250 260

3
Sequenze identiche al 62%

4
Alcune regioni delle proteine sopportano meglio le mutazioni rispetto ad altre. In particolare le regioni importanti per la produzione della struttura secondaria (alfa eliche e foglietti beta) sono poco tolleranti, mentre le regioni di giunzione possono essere molto più variabili.
 sono poco tolleranti, mentre le regioni di giunzione possono essere molto più variabili..")
5
L’evoluzione non opera direttamente né sulla sequenza del DNA né sulla struttura primaria delle proteine, ma sulla conformazione tridimensionale di queste ultime. In considerazione di questo e della degenerazione del codice genetico, la struttura tridimensionale delle proteine è più conservata della sequenza primaria, che a sua volta è più conservata della sequenza dei nucleotidi codificanti. -ATGTTGAAGTTT- - M L K F - -ATGTTGAAGTTT- - M L K F - -ATGTTGAAGTTT- - M L K F - -ATGTTGAAGTTC- - M L K F - Sequenza a.a identica -ATGTTGAAGTAT- - M L K Y - Sequenza a.a diversa, stuttura conservata -ATGTTGAAGGTT- - M L K V - Sequenza a.a diversa, stuttura destabilizzata

6
Identità ed omologia Gli aminoacidi possono essere raggruppati in base alle loro caratteristiche fisico-chimiche. Su questa base un aminoacido può essere definito simile ad un altro Dato un allineamento di sequenze: Percentuale di identità= di residui identici/residui totali*100 (residui identici + residui simili) Percentuale di omologia= * 100 (residui totali) RKRK Carichi positivamente R K D E Carichi F L I V W Idrofobici
 Percentuale di omologia= * 100 (residui totali) RKRK Carichi positivamente R K D E Carichi F L I V W Idrofobici.")
7
ATA --------SSGGYRKGVTEAKLKVAINGFGRIGRNFLRCWHGRKDSPLDIIAIND-TGGV 99 ATB AQIIPKAVTTSTPVRGETVAKLKVAINGFGRIGRNFLRCWHGRKDSPLEVVVLND-SGGV 119 HS ----------------MG--KVKVGVNGFGRIGRLVTRAAF--NSGKVDIVAINDPFIDL 40 MM --------------------MVKVGVNGFGRIGRLVTRAAI--CSGKVEIVAINDPFIDL 38 XL --------------------MVKVGINGFGCIGRLVTRAAF--DSGKVQVVAINDPFIDL 38 DM --------------------MSKIGINGFGRIGRLVLRAAI--DKG-ANVVAVNDPFIDV 37 CE ----------------MS--KANVGINGFGRIGRLVLRAAV--EKDTVQVVAVNDPFITI 40 SP ----------------MA--IPKVGINGFGRIGRIVLRNAI--LTGKIQVVAVNDPFIDL 40 ATC ----------------MADKKIRIGINGFGRIGRLVARVVL--QRDDVELVAVNDPFITT 42 OS ----------------MG--KIKIGINGFGRIGRLVARVAL--QSEDVELVAVNDPFITT 40 SC --------------------MVRVAINGFGRIGRLVMRIAL--SRPNVEVVALNDPFITN 38 ECA -------------------MTIKVGINGFGRIGRIVFRAAQ--KRSDIEIVAIND-LLDA 38 HI -------------------MAIKIGINGFGRIGRIVFRAAQ--HRDDIEVVGIND-LIDV 38 ECC --------------------MSKVGINGFGRIGRLVLRRLL-EVKSNIDVVAIND-LTSP 38.:.:**** ***. * ::: :**. ATA KQASHLLKYDSTLGIFDADVKPSGETAISVD-----GKIIQVVSNRNPSLLPWKELGIDI 154 ATB KNASHLLKYDSMLGTFKAEVKIVDNETISVD-----GKLIKVVSNRDPLKLPWAELGIDI 174 HS NYMVYMFQYDSTHGKFHG-TVKAENGKLVIN-----GNPITIFQERDPSKIKWGDAGAEY 94 MM NYMVYMFQYDSTHGKFNG-TVKAENGKLVIN-----GKPITIFQERDPTNIKWGEAGAEY 92 XL DYMVYMFKYDSTHGRFKG-TVKAENGKLIIN-----DQVITVFQERDPSSIKWGDAGAVY 92 DM NYMVYLFKFDSTHGRFKG-TVAAEGGFLVVN-----GQKITVFSERDPANINWASAGAEY 91 CE DYMVYLFKYDSTHGQFKG-TVTYDGDFLIVQKDGKSSHKIKVFNSKDPAAIAWGSVKADF 99 SP DYMAYMFKYDSTHGRFEG-SVETKGGKLVID-----GHSIDVHNERDPANIKWSASGAEY 94 ATC EYMTYMFKYDSVHGQWKHNELKIKDEKTLLFG----EKPVTVFGIRNPEDIPWAEAGADY 98 OS DYMTYMFKYDTVHGQWKHSDIKIKDSKTLLLG----EKPVTVFGIRNPDEIPWAEAGAEY 96 SC DYAAYMFKYDSTHGRYAG-EVSHDDKHIIVD-----GKKIATYQERDPANLPWGSSNVDI 92 ECA DYMAYMLKYDSTHGRFDG-TVEVKDGHLIVN-----GKKIRVTAERDPANLKWDEVGVDV 92 HI EYMAYMLKYDSTHGRFDG-TVEVKDGNLVVN-----GKTIRVTAERDPANLNWGAIGVDI 92 ECC KILAYLLKHDSNYGPFPW-SVDFTEDSLIVD-----GKSIAVYAEKEAKNIPWKAKGAEI 92. ::::.*: * : : : : ::. : * Allineamento multiplo

8
Calcolo matrici di sostituzione su base evolutiva KQASHLLKYDSTLGIFDADVKPSGETAISVD KQASHLVKYDSTLGIFDADVRPSGETAISVD KQASHLLKYESTLGIFDADVKPSGATAISVD KQACHLLKYDSTLGIFDAEVKPSGETAISVD KQASHLLKYDSTLGIFDADVKPSGETAISVD KQASHLLKYDSSLGIFDADVKASGETAISVD KQASHILKYDSTLGIFDADVKPSGETAITVD DQASHLLKYDSTLGLFDADVKPSGETAISVD KQASHLLKFDSTLGIFDADVRPSGETAISVD KQASSLLKYDSTLGIFDADVKPTGETAISVD KQVSHLLKYDSTLGIFEADVKPSGETAISVD KQASHLLKYDVTLGIFDADVKPSGETPISVD KQASHLCKYDSTLGIFDADVKPSVETAISVD KQASHLLKYQSTLGIFDADVKPSGETAISVE Freq. Oss. Coppia a.a. Odd= Freq. attesa Coppia a.a. Score= ln Odd (bit)
.")
9
Matrici di sostituzione Serie PAM (percent accepted mutation) Il presupposto per l’elaborazione di questo tipo di matrici è che in sequenze correllate le sostituzioni di aminoacidi che si osservano non disturbano molto la struttura della proteina, e sono quindi accettate dall’evoluzione. Allineamento multiplo di sequenze poco divergenti (1%) Misurazione della frequenza con cui un aminoacido veniva cambiato in un altro, per tutte le possibili coppie di aminoacidi Calcolo del log del rapporto tra la frequenza di una determinata sostituzione e la frequenza attesa in base al caso In questo modo è stata ottenuta la matrice PAM1
 Misurazione della frequenza con cui un aminoacido veniva cambiato in un altro, per tutte le possibili coppie di aminoacidi Calcolo del log del rapporto tra la frequenza di una determinata sostituzione e la frequenza attesa in base al caso In questo modo è stata ottenuta la matrice PAM1.")
10
Matrici di sostituzione Serie PAM (percent accepted mutation) 1 PAM equivale a circa 10 milioni di anni di divergenza Dalla matrice PAM 1 sono state ottenute altre matrici per estrapolazione a distanze evolutive maggiori. Le matrici PAM si sono rivelate molto utili, anche se corrispondono a frequenze di sostituzione derivate piuttosto che osservate direttamente. Le matrici a basso PAM sono migliori per confrontare sequenze molto simili o molto corte. Quelle ad alto PAM sono ottimali per sequenze più divergenti. In base a simulazioni effettuate le matrici PAM120, PAM80 e PAM60 sono ottimali per sequenze che mostrano il 40%, 50% e 60% di similarità, rispettivamente.

11
Matrici di sostituzione Serie BLOSUM (Blocks Amino Acid Substitution Matrices) La metodica statistica utilizzata è la stessa che per le matrici PAM. La differenza è che le variazioni degli aminoacidi sono state direttamente misurate su almeno 250 blocchi di aminoacidi privi di gaps, conservati in proteine anche molto divergenti. In questo caso i numeri stanno ad indicare la percentuale di identità tra i blocchi che hanno contribuito alla costruzione della matrice. Ne consegue che le matrici ad alto BLOSUM (ad es. 80) sono indicate per confrontare proteine meno divergenti, mentre quelle a basso blosum (30 o 40) sono adatte per proteine più divergenti. Il vantaggio principale di queste matrici è che le frequenze di sostituzione non sono estrapolate ma misurate direttamente.
 sono indicate per confrontare proteine meno divergenti, mentre quelle a basso blosum (30 o 40) sono adatte per proteine più divergenti. Il vantaggio principale di queste matrici è che le frequenze di sostituzione non sono estrapolate ma misurate direttamente..")
12
Matrice PAM 30

13
Scoring matrix = PAM 30 Gap open penalty = -10 Gap extension penalty = -2 Calcolo dello score di un allineamento

14
* * * * * * * * * * ** * * * * * * P A O L O R O S S I PAOLOROSSIPAOLOROSSI Comparazione di sequenze: Dot Plot Analysis Sequenze identiche

15
* * * * * * * * * * * * * * * P A O L O R O S S I PAOLARUSSOPAOLARUSSO Comparazione di sequenze: Dot Plot Analysis Sequenze simili

16
* * * * * * P A O L O R O S S I CARLAVERDICARLAVERDI Comparazione di sequenze: Dot Plot Analysis Sequenze diverse

17
P A O L O M A R I A R O S S I PAOLOROSSIPAOLOROSSI Comparazione di sequenze: Dot Plot Analysis * *** *** * *** ** *** ** ** * Inserzioni/delezioni

18
7-6-9-8-15-8-2 -69 -2-70 -6 -9-670-12-7-8 -4 -10-152-3-12 -15-7-12-213-8-13-6 -80-6-88-4-7 -6 -4-3-500-8 0-6-88-4-7 -3 -2-13-3-4-5 2-9-6-5-14-5 V H K R W N A L L L Comparazione di sequenze: Dot Plot Analysis VHKDWNSNQIVHKDWNSNQI

19
7-6-9-8-15-8-2 -69 -2-70 -6 -9-670-12-7-8 -4 -10-152-3-12 -15-7-12-213-8-13-6 -80-6-88-4-7 -6 -4-3-500-8 0-6-88-4-7 -3 -2-13-3-4-5 2-9-6-5-14-5 V H K R W N A L L L Comparazione di sequenze: Dot Plot Analysis VHKDWNSNQIVHKDWNSNQI

20
Allineamento globale e allineamento locale Alcuni programmi, date due sequenze, generano in ogni caso l’allineamento migliore possibile tra di esse, su tutta la lunghezza. Questo tipo di allineamento prende il nome di allineamento globale. Non necessariamente un allineamento globale ha significato biologico. Altri programmi, date due sequenze, non le allineano necessariamente su tutta la lunghezza, ma vanno a cercare soltanto i tratti in cui l’omologia tra le due sequenze (ossia lo score) supera una certa soglia. In questi si parla di allineamento locale.
 supera una certa soglia. In questi si parla di allineamento locale..")
21
Alcuni dei programmi più utilizzati Algoritmo di Needleman-Wunsch = allineamento globale, matematicamente rigoroso. Molto lento mai utilizzato per ricerhe in banca dati. Algoritmo di Smith-Watermann = allineamento locale, matematicamente rigoroso. Molto lento, utilizzato solo recentemente per ricerhe in banca dati, grazie allo sviluppo di calcolatori dedicati. FASTA = allineamento locale, piuttosto rigoroso, più veloce dei precedenti. BLAST = (Basic Local Alignment Search Tool). Poco rigoroso, ma estremamente più veloce dei precedenti. Pertanto è molto utilizzato per le ricerche di routine. Gli altri sono più utilzzati quando si vogliono trovare deboli similitudini.
. Poco rigoroso, ma estremamente più veloce dei precedenti. Pertanto è molto utilizzato per le ricerche di routine. Gli altri sono più utilzzati quando si vogliono trovare deboli similitudini..")
22
Come funziona BLAST? La sequenza di cui si vogliono trovare gli omologhi viene scomposta in tutte le possibili parole di una lunghezza prefissata (ad esempio due o tre se si tratta di aminoacidi) MLFFRRQPKHCSDTEF MLFLFFFFRFRRRRQRQPQPK PKHKHCHCSCSDSDTDTETEF Le parole di tre lettere vengono quindi cercate in tutte le sequenze della banca dati (operazione estremamente rapida)
 MLFFRRQPKHCSDTEF MLFLFFFFRFRRRRQRQPQPK PKHKHCHCSCSDSDTDTETEF Le parole di tre lettere vengono quindi cercate in tutte le sequenze della banca dati (operazione estremamente rapida).")
23
Query: MLFFRRQPKHCSDTEF MLFLFFFFRFRRRRQRQPQPK PKHKHCHCSCSDSDTDTETEF Subject: ASDDERTGLFDRKQPKACMDSEFKATT QPK ::: In seguito l’allineamento viene esteso HCSDTEF : :.: MLFFRR :: :. Subject: ASDDERTGLFDRKQPKACMDSEFKATT QPK ::: Query:

24
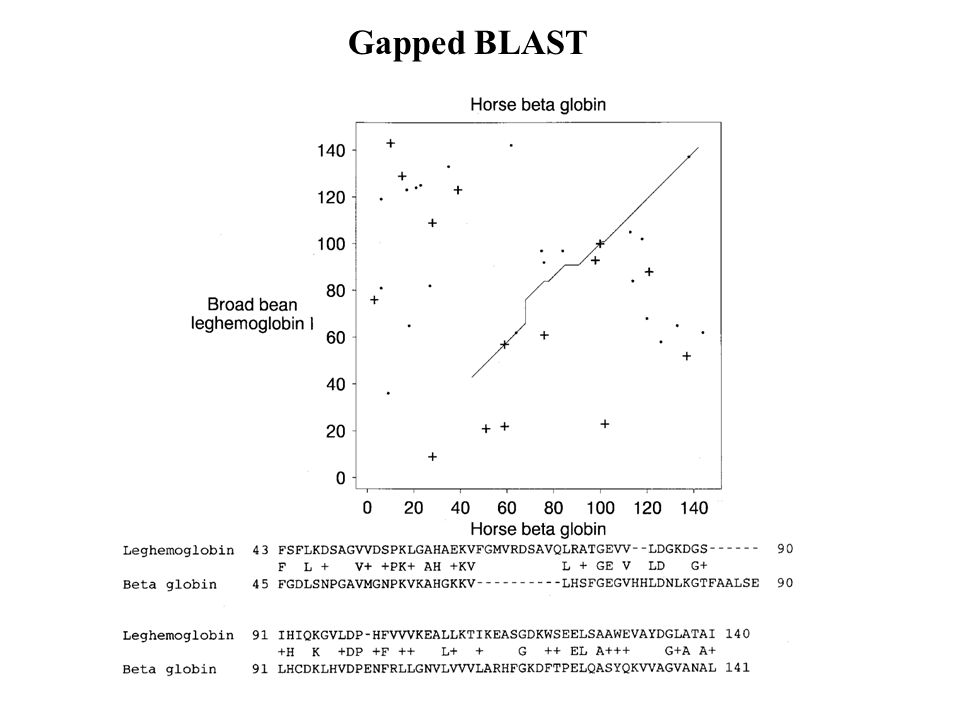
Gapped BLAST

26
Versioni disponibili del programma Blast BlastN = ricerca in un database di sequenze nucleotidiche le sequenze omologhe ad una sequenza nucleotidica data. BlastP = ricerca in un database di sequenze aminoacidiche le sequenze omologhe ad una sequenza aminoacidica data. BlastX = data una sequenza nucleotidica, la traduce in tutte e sei le possibili cornici di lettura (traduzione dinamica), e cerca le seqeunze omologhe alle traduzioni in un database di sequenze aminoacidiche tBlastN = data una sequenza aminoacidica, la confronta con tutte le possibili traduzioni di un database di sequenze nucleotidiche. tBlastX = data una sequenza nucleotidica, confronta la sua traduzione dinamica con tutte le possibili traduzioni di un database di sequenze nucleotidiche.
, e cerca le seqeunze omologhe alle traduzioni in un database di sequenze aminoacidiche tBlastN = data una sequenza aminoacidica, la confronta con tutte le possibili traduzioni di un database di sequenze nucleotidiche. tBlastX = data una sequenza nucleotidica, confronta la sua traduzione dinamica con tutte le possibili traduzioni di un database di sequenze nucleotidiche..")
27
Database più utilizzati NR = non ridondante, sia nucleotidico che aminoacidico. Contiene numerosissime annotazioni funzionali. Database di prima scelta, qualunque sequenza si stia studiando. EST = unicamente nucleotidico, con pochissima annotazione e costituito da sequenze di bassa qualità, ma preziosissimo per molte applicazioni, come lo studio di geni non precedentemente caratterizzati Database genomici dei diversi organismi = alcuni sono unicamente nucleotidici, ma in genere esiste anche una versione delle sequenze aminoacidiche identificate o predette. PDB = database delle strutture atomiche SwissProt = aminoacidico, è in assoluto quello che contiene più informazioni sulle proteine

28
Geni ortologhi Due geni appartenenti a specie diverse si definiscono ortologhi se hanno una forte omologia e svolgono la stessa funzione. Quasi certamente le proteine codificate hanno strutture tridimensionali sovrapponibili. Ùquesti geni hanno avuto origine da un antenato comune in un periodo evolutivo precedente. Geni paraloghi Si definiscono paraloghi i geni derivanti da eventi di duplicazione e divergenza nel genoma della stessa specie. Questi eventi possono portare alla produzione di famiglie di proteine correlate con strutture e funzioni biologiche simili. A volte però i paraloghi possono diversificarsi moltissimo, ed acquisire funzioni specializzate nonostante la struttura simile.

29
Programma per Dot Plot Analysis: DOTTER http://www.cgr.ki.se/cgr/groups/ sonnhammer/Dotter.html

30
Esercizio 1 Produrre un dot plot delle sequenze k1nt.txt e k2nt.txt Esercizio 2 Produrre un dot plot delle sequenze k1aa.txt e k2aa.txt

31
Esercizio 3 Produrre un dot plot della sequenza notch.txt contro se stessa Esercizio 4 Produrre un dot plot delle sequenze notch.txt e lin12.txt

32
Allineamento globale Esercizio 5: Importare le sequenze notch.txt e lin12.txt in Biology Workbench (http://workbench.sdsc.edu) Produrre un allineamento delle due sequenze con il programma ALIGN (parametri di default)
")
36
Allineamento globale Esercizio 6: Produrre un allineamento delle stesse sequenze con il programma ALIGN utilizzando la matrice PAM250 invece della Blosum50. Si ottiene lo stesso allineamento ottimale?

37
Allineamento locale Esercizio 7: Produrre un allineamento delle stesse sequenze con il programma LALIGN. Che cosa è cambiato?

38
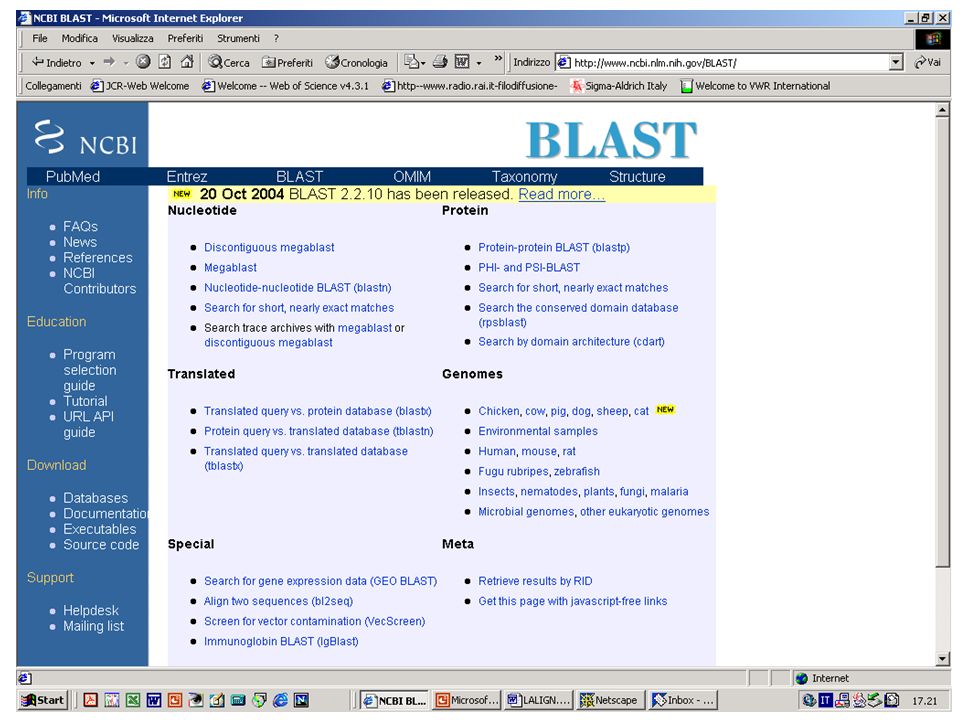
BLAST http://www.ncbi.nlm.nih.gov/blast/index.html http://www.ch.embnet.org/software/aBLAST.html 130.192.70.75

39
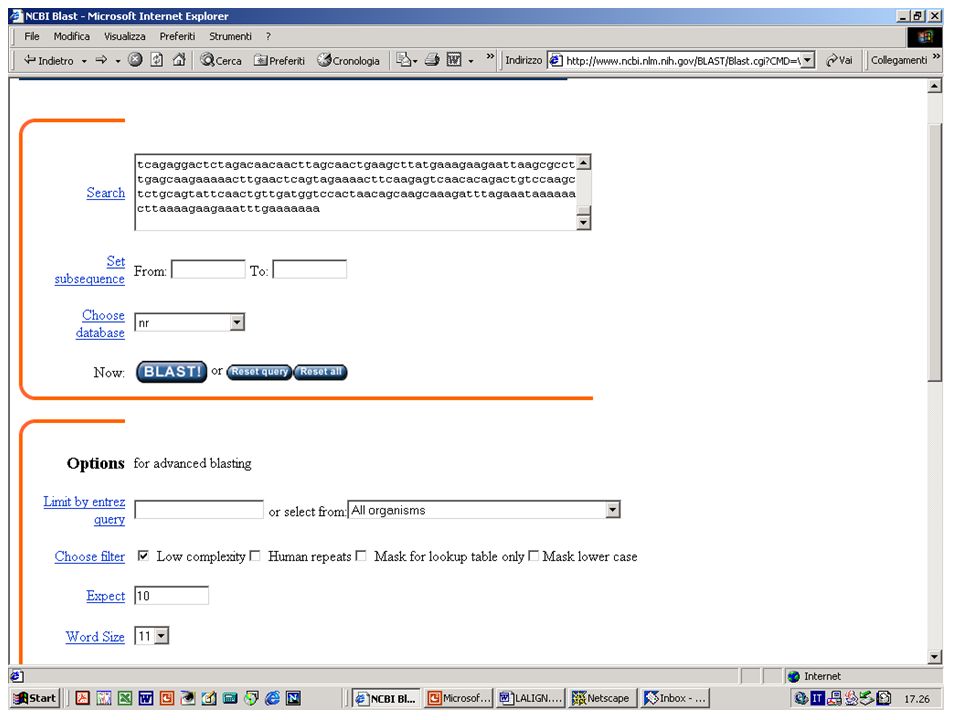
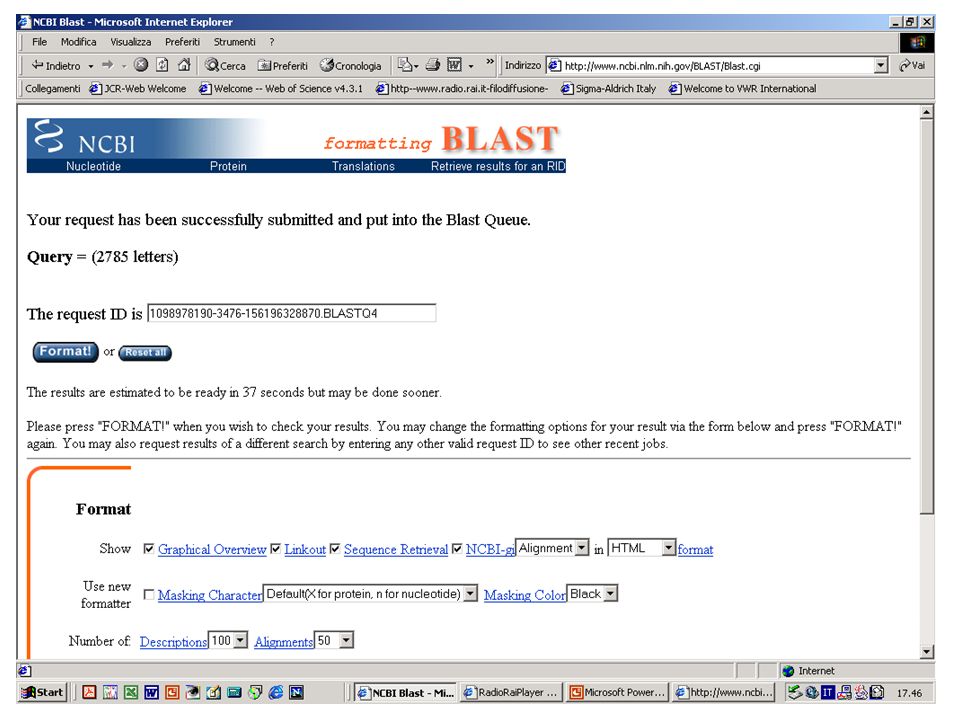
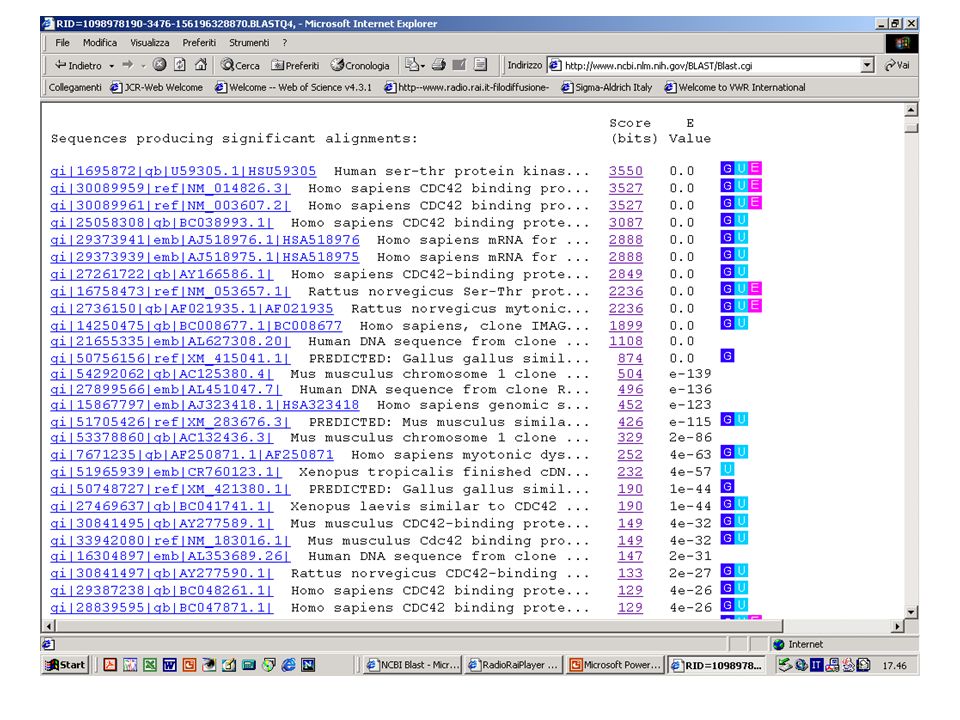
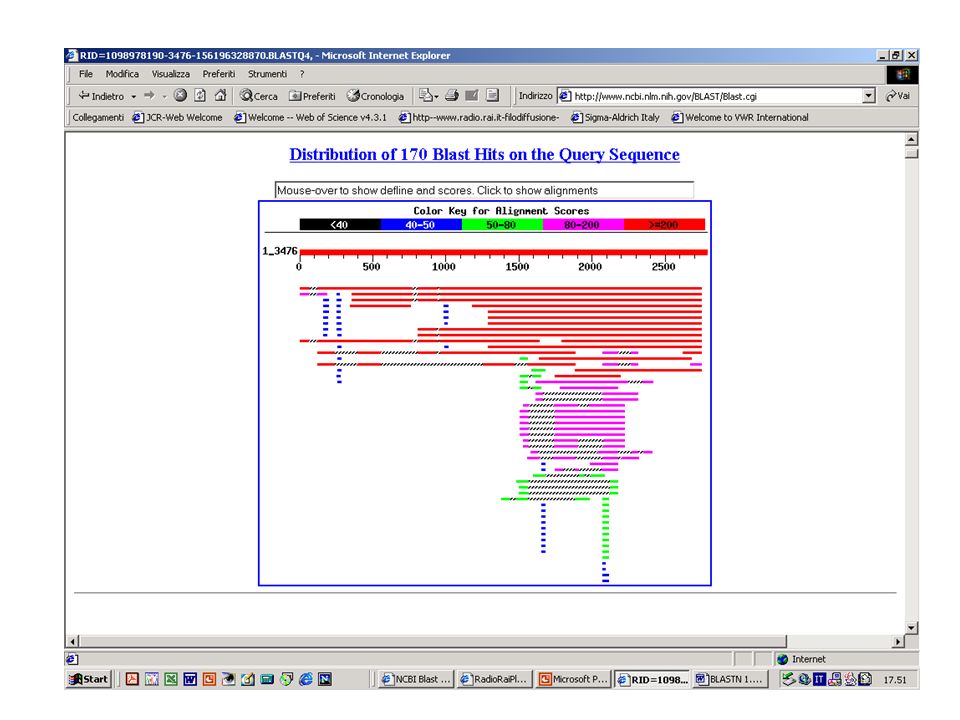
BlastN, BlastX o BlastP? Esercizio 8 Eseguire una ricerca con BlastN su database NR con la sequenza k2nt.txt.

45
Ricerca senza filtro per le regioni a bassa complessità

46
BlastN, BlastX o BlastP? Esercizio 9 Eseguire una ricerca con BlastX su database NR con la stessa sequenza.

48
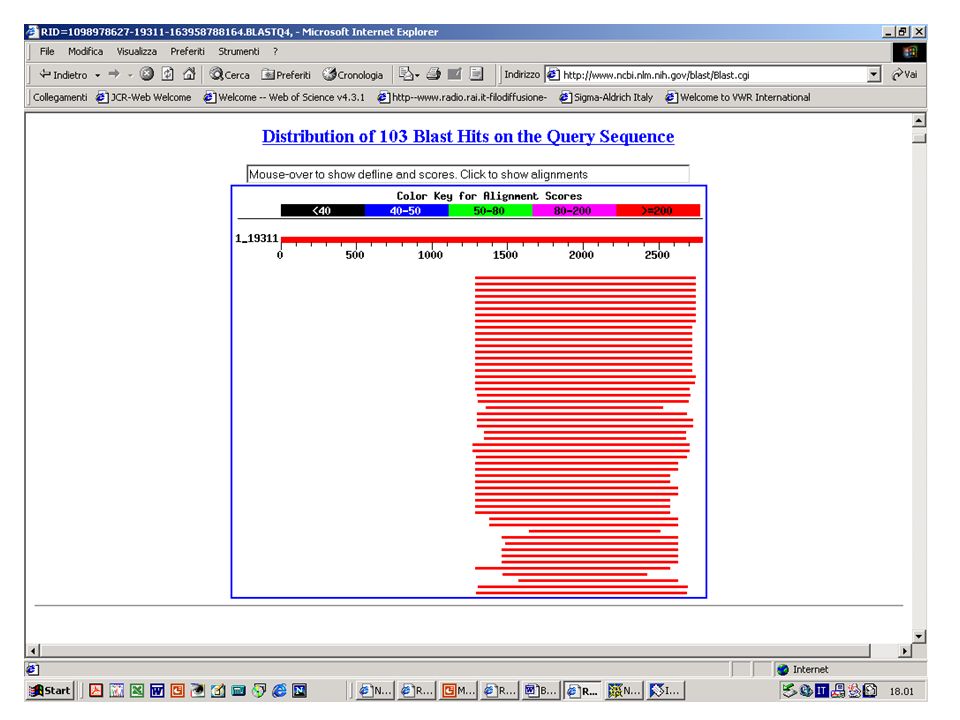
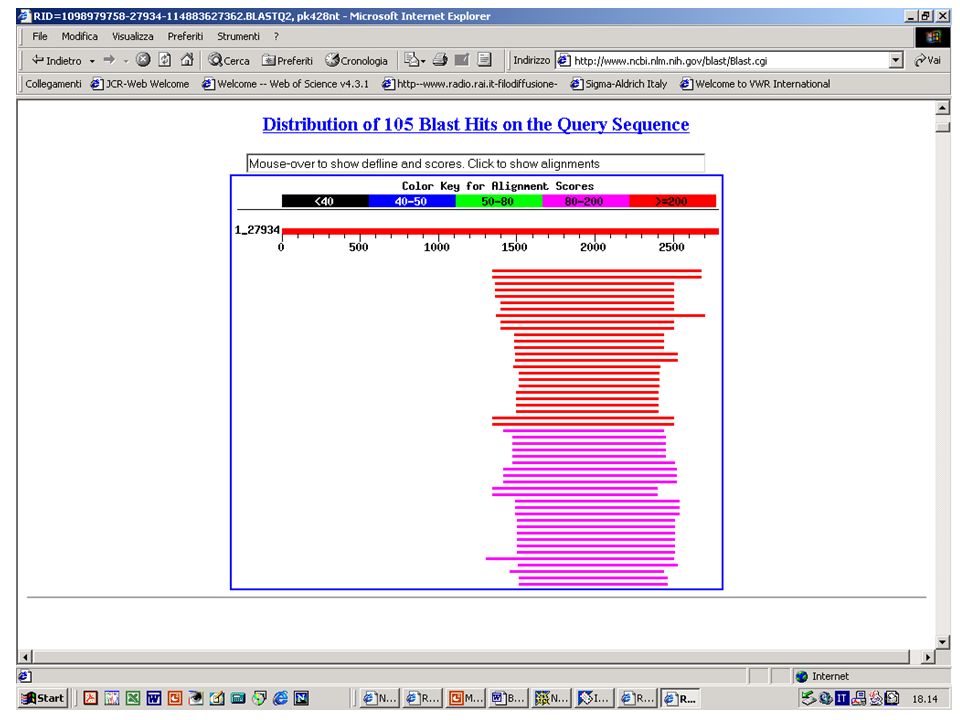
BlastN, BlastX o BlastP? Esercizio 10 Eseguire una ricerca con BlastN sul database NR selezionando come organismo ‘Drosophila Melanogaster’ con la sequenza k2nt.txt.

51
BlastN, BlastX o BlastP? Esercizio 11 Eseguire una ricerca con BlastX sul database NR selezionando come organismo ‘Drosophila Melanogaster’ con la sequenza k2nt.txt.

55
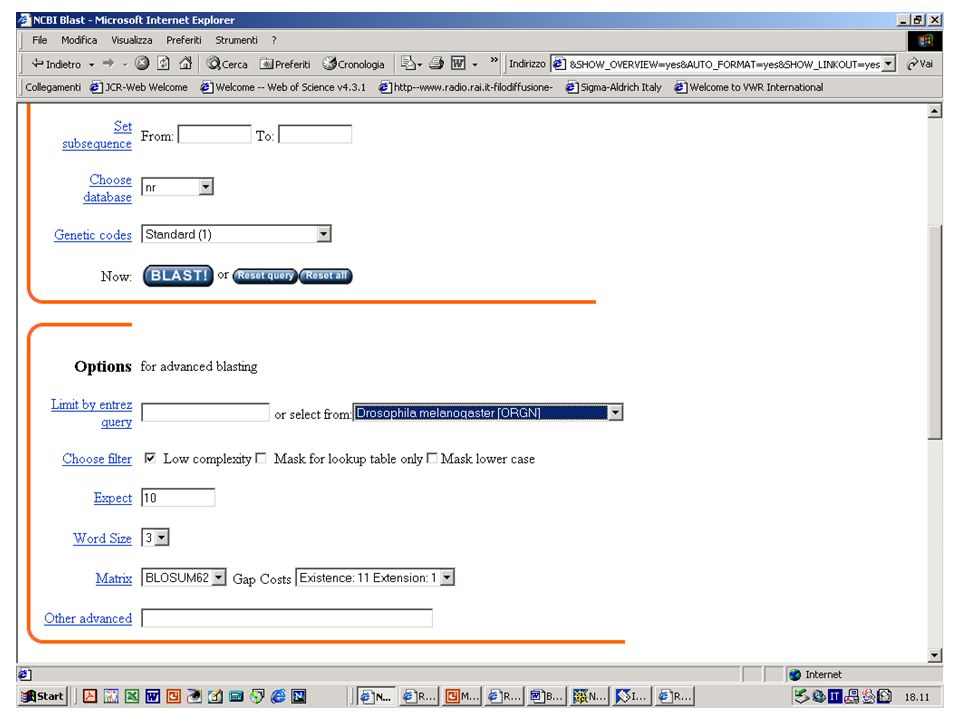
Filtri di BLAST Esercizio 12 Eseguire una ricerca con BlastP su database NR con la sequenza NCT.txt. Esercizio 13 Eseguire una ricerca con le stesse impostazioni ma eliminando il filtro per le regioni low complexity.

56
Con filtro

57
Senza filtro

58
http://repeatmasker.genome.washington.edu /cgi-bin/RepeatMasker RepeatMasker

59
Importanza della lunghezza della query E(S)=Kmne - S m= grandezza del database n=lunghezza della sequenza S=score
=Kmne - S m= grandezza del database n=lunghezza della sequenza S=score")
60
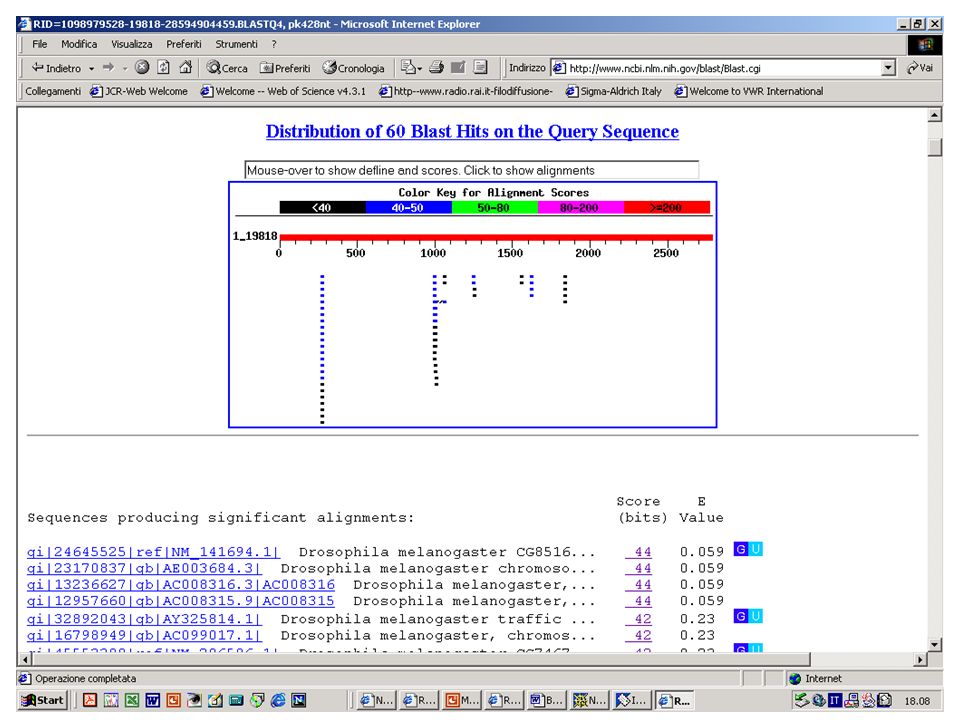
Importanza della lunghezza Esercizio 14 Eseguire una ricerca con BlastP su database NR con la sequenza: MSGEVRLRQLEQFILDGP Esercizio 15 Rieseguire la ricerca con gli stessi parametri e con la sequenza: MSGEVRLRQLEQFILDGPAQTNGQYFSVETLLDILIC LYDECNN Come si modificano lo score e il valore E?

63
Importanza della matrice Esercizio 12 Eseguire una ricerca con BlastP su database NR con la sequenza: MSGEVRLRQLEQFILDGP Esercizio 13 Eseguire la stessa ricerca cambiando la matrice da BLOSUM62 prima a PAM30 e poi a PAM 250. Come si modificano i risultati? Perché nel primo caso trovo più sequenze e nel secondo meno?

64
BLOSUM62

65
PAM250

66
PAM30

67
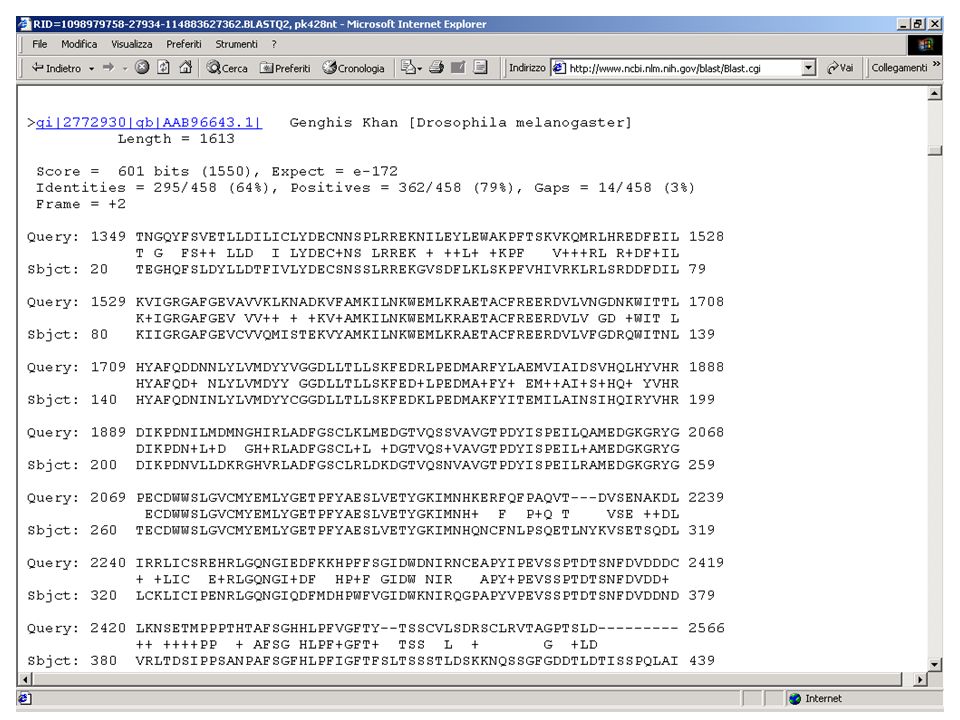
Una volta stabilito che un insieme di proteine sono tra di loro omologhe posso procedere ad un allineamento multiplo. Il programma più usato a questo scopo si chiama CLUSTALW. Da un allineamento multiplo posso derivare molte informazioni. In particolare mi può aiutare a fare una predizione di struttura secondaria, a dire quali sono gli aminoacidi essenziali per tutta la famiglia, a dire quali sono gli aminoacidi che conferiscono particolari caratteristiche, a identificare particolari domini funzionali.

68
CLUSTALW

69
Esercizio 1 Utilizzando il programma CLUSTALW, generare un allineamento multiplo delle sequenze contenute nel file GAPDH.TXT ftp://ftp.ebi.ac.uk/pub/software/dos/clustalw/ http://www.ebi.ac.uk/clustalw/ Esercizio 2 Visualizzare i risultati con il programma Jalview: http://www.ebi.ac.uk/~michele/jalview/contents.html

Presentazioni simili




 mettere in discussione l’idea del progetto 2) rivalutare il.>")











 Algoritmi di allineamento 2) Algoritmi di ricerca in database>")




 BLAST: Altshul (1990)>")