Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
In questa lezione ci occuperemo
Ricerca di pattern e motivi funzionali in sequenze nucleotidiche Ricerca di pattern e motivi funzionali in sequenze proteiche Potremmo enunciare il seguente paradigma: La sequenza del DNA determina la sequenza di una proteina La sequenza di una proteina ne determina la struttura La struttura di una proteina ne determina la funzione

2
Dal DNA …………….. alle Proteine

3
Sequenze nucleotidiche Sequenze proteiche
TRADUZIONE

4
… ctggcccacaagtatcactac…
Esercizio relativo alla traduzione di una sequenza nucleotidica in una sequenza amminoacidica Data la sequenza del gene della b-emoglobina umana: … ctggcccacaagtatcactac… 1)Scrivere la traduzione di questa sequenza in una sequenza amminoacidica 2)Scrivere la sequenza nucleotidica per un cambiamento di una singola base che produca una mutazione silente in questa regione (la mutazione silete è quella che lascia invariata la sequenza amminoacidica) 3)Scrivere la sequenza nucleotidica e la traduzione in sequenza amminoacidica per un cambiamento di una singola base che produca una mutazione di un amminoacido.
Scrivere la traduzione di questa sequenza in una sequenza amminoacidica. 2)Scrivere la sequenza nucleotidica per un cambiamento di una singola base che produca una mutazione silente in questa regione (la mutazione silete è quella che lascia invariata la sequenza amminoacidica) 3)Scrivere la sequenza nucleotidica e la traduzione in sequenza amminoacidica per un cambiamento di una singola base che produca una mutazione di un amminoacido.")
5
Ricerca di pattern e di motivi funzionali
Qualche definizione necessaria ……. Un motivo di interesse biologico è costituito da un insieme di caratteri (nucleotidi o amminoacidi) non necessariamente contigui nella sequenza ma che si trovano sempre o sono spesso associati ad una precisa struttura e funzione biologica (ad esempio: promotori o hanno la stessa capacità di legare nucleotidi) La bioinformatica si occupa di sviluppare metodi per il riconoscimento di pattern di interesse biologico e di curare banche dati in cui tali pattern siano organizzati e resi disponibili per l’analisi strutturale e funzionale di nuove sequenze. Ciò deriva dal fatto che nel corso dell’evoluzione la natura ha sviluppato uno o pochi modi per erealizzare una nuova funzione (ad es. attività catalitica o altro)
 non necessariamente contigui nella sequenza ma che si trovano sempre o sono spesso associati ad una precisa struttura e funzione biologica (ad esempio: promotori o hanno la stessa capacità di legare nucleotidi) La bioinformatica si occupa di sviluppare metodi per il riconoscimento di pattern di interesse biologico e di curare banche dati in cui tali pattern siano organizzati e resi disponibili per l’analisi strutturale e funzionale di nuove sequenze. Ciò deriva dal fatto che nel corso dell’evoluzione la natura ha sviluppato uno o pochi modi per erealizzare una nuova funzione (ad es. attività catalitica o altro)")
6
Per quanto riguarda la Ricerca di pattern e motivi funzionali in sequenze nucleotidiche
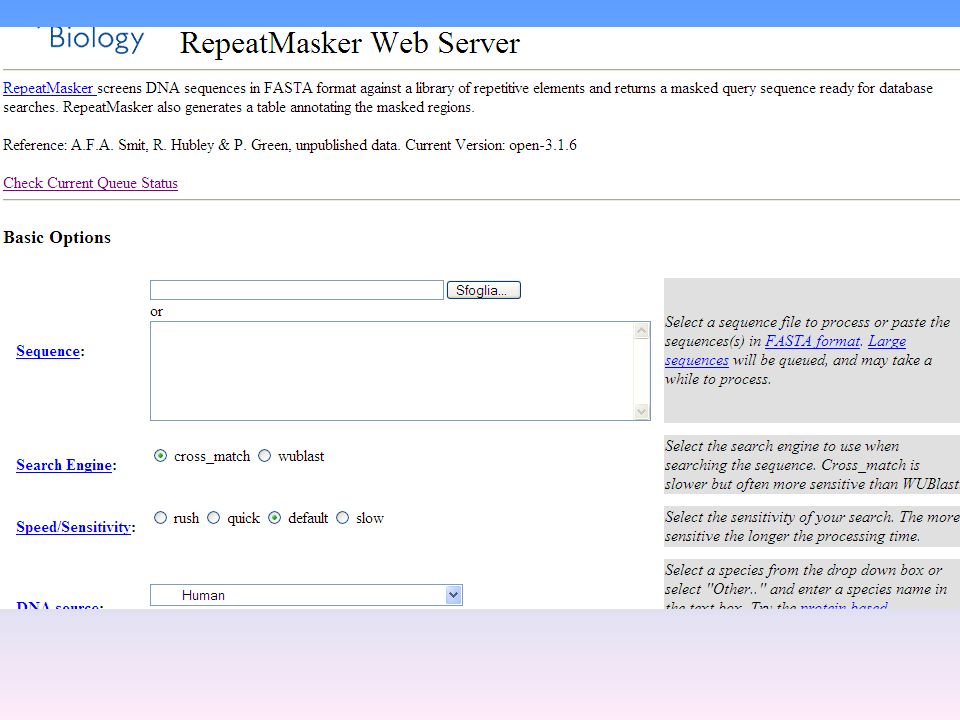
In realtà non ci sono strumenti che possono essere utilizzati indifferentemente per l’analisi di una qualsiasi sequenza nucleotidica. Infatti nella studio dei segnali importanti per l’identificazione dei singoli geni è necessario considerare alcuni punti: Alcuni programmi sono stati sviluppati per un organismo specifico o per un numero limitato di organismi Per tutte le sequenze è necessario un filtro che escluda dall’analisi le sequenze ripetitive. Infatti, una grande parte del DNA è costituito da sequenze di DNA ripetute che non fanno parte di regioni codificanti. Queste sequenze devono essere eliminate perché possono interferire con le misure di similarità biologicamente significative nel corso delle ricerche in banche dati Ci sono due programmi che fanno questo: CENSOR ( e RepeatMasker ( Questi due programmi accedono a raccolte di sequenze di DNA ripetute ed operano un confronto con le sequenze sottomesse al programma riuscendo ad identificare le sequenze ripetute presenti e le sottraggono dalla ricerca.

7
Censor può essere usato con sequenze proteiche e nucleotidiche
Possiamo scegliere un organismo per il quale fare la ricerca Scegliere di andare a valutare le percentuali di identità e non di similarità come viene fatta di default

9
A noi potrebbe interessare:
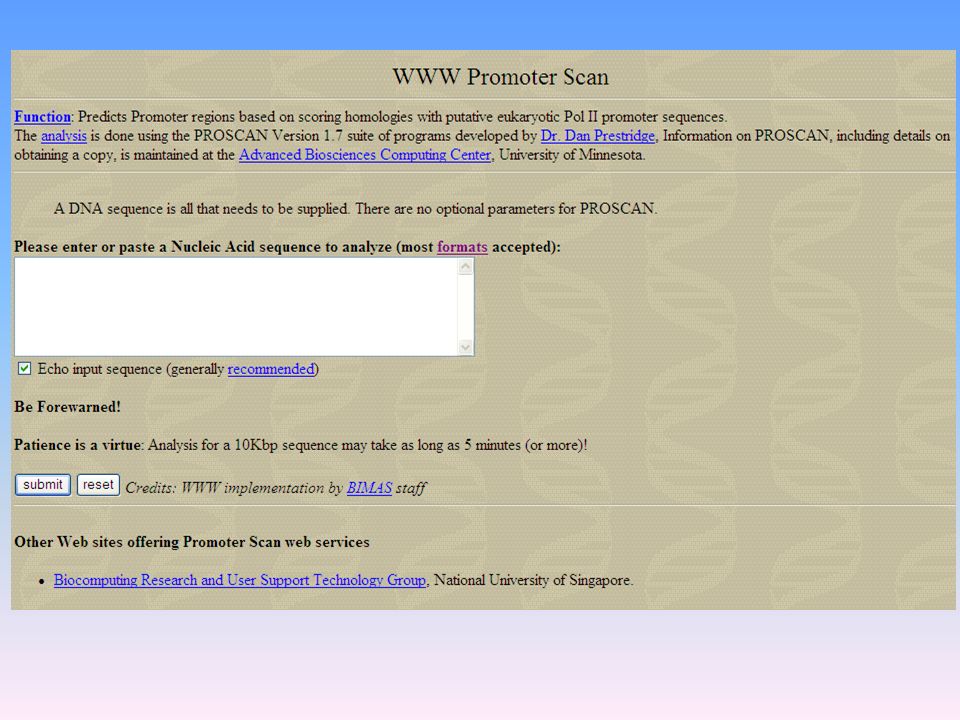
Ricercare i promotori eucariotici L’identificazione dei promotori è importante per l’identificazione di sequenze geniche codificanti e per la corretta assegnazione di esoni tra i geni situati nella stessa porzione del cromosoma (PromoterScan: 2) Ricercare i siti di giunzione tra introni ed esoni Un gene è costituito da una sequenza codificante interrotta da sequenze non codificanti (dette introni). I geni sono combinazioni di corti esoni ed introni di lunghezza variabile. Il termine esoni si applica a tutte le regioni che non sono eliminate nel corso di maturazione del RNA [cioè le regioni non tradotte al 5’ dei geni, quelle codificanti vere e proprie (CDS) e le regioni non tradotte al 3’]. Identificare i siti di giunzione tra introni ed esoni per una corretta predizione della struttura di un gene. NetGene: GenScan: GenScan è il programma più usato per predire la struttura di un gene
 Ricercare i siti di giunzione tra introni ed esoni. Un gene è costituito da una sequenza codificante interrotta da sequenze non codificanti (dette introni). I geni sono combinazioni di corti esoni ed introni di lunghezza variabile. Il termine esoni si applica a tutte le regioni che non sono eliminate nel corso di maturazione del RNA [cioè le regioni non tradotte al 5’ dei geni, quelle codificanti vere e proprie (CDS) e le regioni non tradotte al 3’]. Identificare i siti di giunzione tra introni ed esoni per una corretta predizione della struttura di un gene. NetGene: GenScan: GenScan è il programma più usato per predire la struttura di un gene.")
10
3) Siti di inizio della traduzione
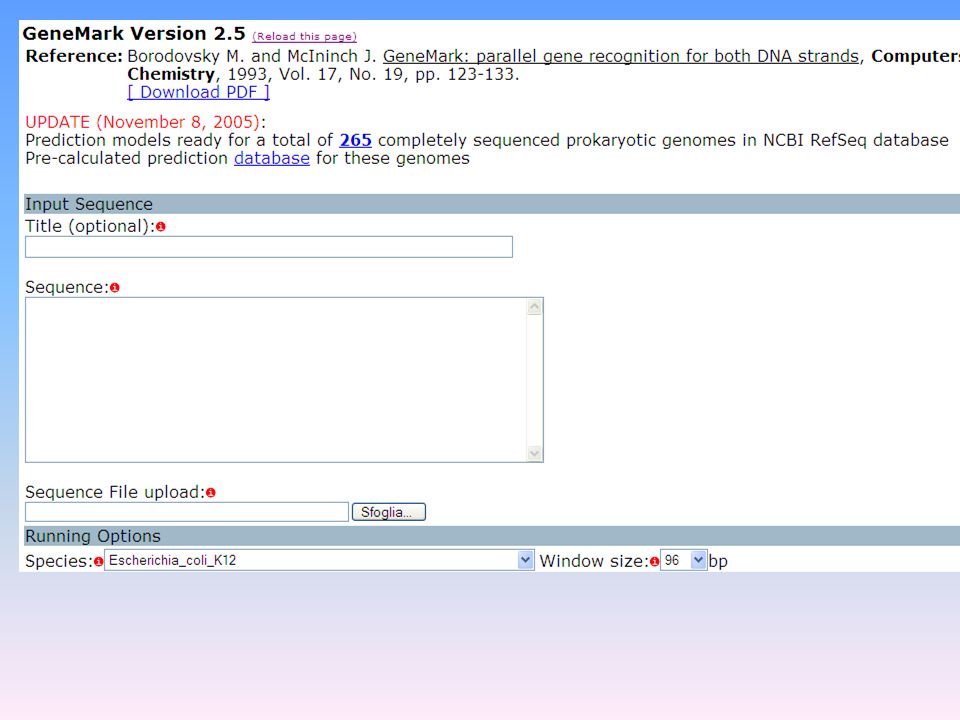
Il codone di inizio è in generale (anche se non sempre) il codone AUG che codifica per la Metionina (GeneMark: 4) Identificazione dei segnali di poliadenilazione e di terminazione della traduzione La più nota sequenza segnale coinvolta nella poliadenilazione è AATAAA (GRAIL:
 il codone AUG che codifica per la Metionina. (GeneMark: 4) Identificazione dei segnali di poliadenilazione e di terminazione della traduzione. La più nota sequenza segnale coinvolta nella poliadenilazione è AATAAA. (GRAIL:")
14
Esercizio 1: Predizioni dei geni codificanti proteine in sequenze genomiche mediante GenScan
Ricerchiamo i geni in una sequenza genomica prodotta nell’ambito del progetto di sequenziamento del genoma di Fugu. Collegandosi al sito: si effettua la ricerca della sequenza scaffold S La sequenza così estratta può essere utilizzata per la predizione utilizzando il programma GenScan. La sequenza estratta viene incollata nella box clicca su Run GenScan Nell’output di GenScan sono indicati tutti i geni predetti, per ciascuno dei quali viene riportata la corrispondente ipotetica sequenza amminoacidica. Queste sequenze possono essere caratterizzati: o effettuando una ricerca con BLAST contro la banca dati delle proteine o ricercando i domini o motivi funzionali attraverso il sistema InterPro

15
Esercizio 2: Caratterizzazione di ipotetiche proteine predette mediante BLAST
Selezionata una proteina dall’entry di GenScan ottenuta nell’esercizio precedente, copiare la sequenza in BLASTP e lanciare la ricerca. Quante proteine omologhe troviamo? Quale proteina è quella che ha una percentuale di identità di sequenza maggiore?

16
Ripetere i tre esercizi precedenti usando lo scaffold S000194
Esercizio 3: Stabilire con precisione la struttura di uno specifico gene usando GenomeScan ( A partire dalla sequenza genomica (S004519) e dalla proteina omologa selezionata con la più alta percentuale di identità di sequenza RunGenomeScan Ripetere i tre esercizi precedenti usando lo scaffold S000194
 e dalla proteina omologa selezionata con la più alta percentuale di identità di sequenza. RunGenomeScan. Ripetere i tre esercizi precedenti usando lo scaffold S")
17
Esercizio 4: Determinazione della struttura di un gene mediante il confronto tra la sequenza genomica e l’mRNA maturo mediante il programma SPIDEY L’allineamento tra una sequenza genomica contenente un gene e la sequenza dell’mRNA corrispondente determina la struttura del gene con l’esatta localizzazione degli introni e degli esoni. Come procedere? Trovare la sequenza genomica di cox4 umano mediante SRS Incollare la sequenza di cox4 (NT_024767) nella box sulla pagina di SPIDEY ed indicare l’accession number della sequenza del trascritto NM_ nel riquadro in basso. Il risultato in SPIDEY mostrerà la struttura di cox4, di esoni ed introni
 nella box sulla pagina di SPIDEY ed indicare l’accession number della sequenza del trascritto NM_ nel riquadro in basso. Il risultato in SPIDEY mostrerà la struttura di cox4, di esoni ed introni.")
18
SPIDEY Potete inserire o le sequenze o gli accession number Quanti esoni avete trovato usando SPIDEY? Ripetere questa stessa ricerca con GenScan usando la stessa sequenza di cox4

19
Ricerca di pattern e di motivi funzionali in sequenze proteiche
Le proteine possono essere raggruppate in un numero limitato di famiglie sulla base della similarità di sequenze. Le proteine ed i domini proteici appartenenti ad una stessa famiglia condividono attributi funzionali e strutturali derivanti da un progenitore comune. Dallo studio di allineamenti multipli di sequenze appartenenti ad una stessa famiglia è evidente che alcune regioni sono più conservate di altre queste regioni conservate sono in generale importanti per la funzione e la struttura di una proteina. Analizzando le regioni costanti e variabili in un allineamento multiplo è possibile identificare un motivo che possa servire alla classificazione funzionale delle proteine che lo contengono.

20
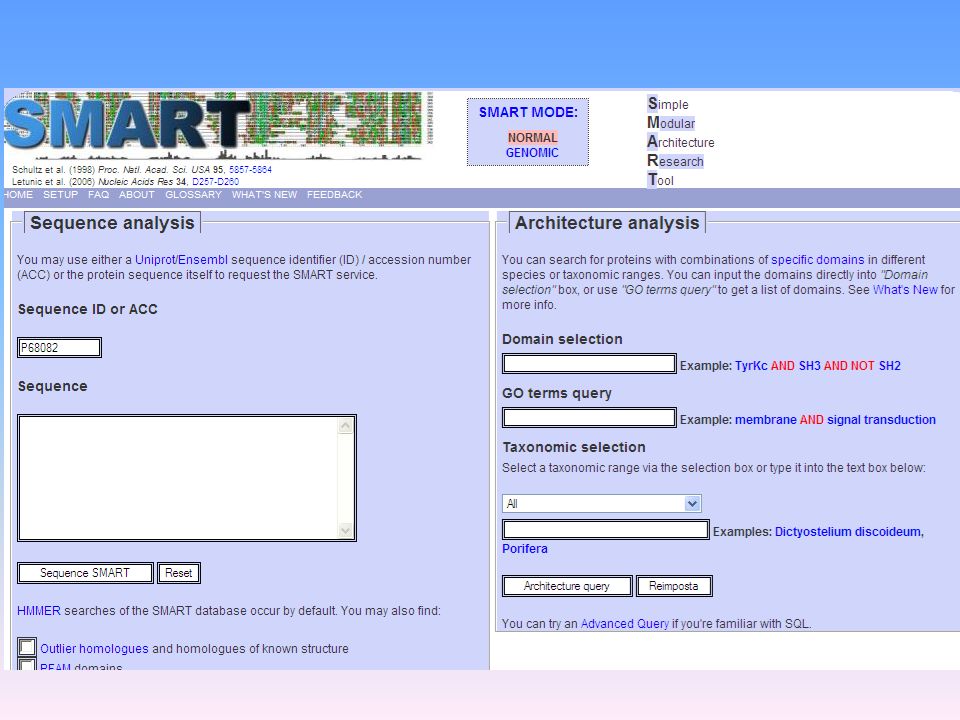
Vari programmi in rete:
Individuazione di domini SMART PFAM Individuazione di motivi funzionali PROSITE PSORT ELM SMART: PFAM

22
[Ala o Gly]-x-x-x-x-GlySer[Ser o Thr]
PROSITE http :// È una banca dati che raccoglie più di 1600 motivi proteici associati ad una determinata struttura e funzione. La sintassi di PROSITE: x indica la posizione in cui ciascun residuo viene accettato Tra le parentesi [ ] sono indicati i residui consentiti in una posizione Tra le parentesi { } sono indicati i residui NON consentiti in una posizione Ad esempio: [A,G]x4GK[S,T] viene tradotto come [Ala o Gly]-x-x-x-x-GlySer[Ser o Thr] Qualche esempio pratico: Ricercare in Prosite la sequenza P68082 Scrivere l’ID dell’entry di Prosite e la famiglia a cui appartiene la proteina Ricercare l’accession number relativo all’interleuchina 1 beta umana (usando ….) e ricercare in Prosite a quale famiglia appartiene questa proteina
![[Ala o Gly]-x-x-x-x-GlySer[Ser o Thr]](http://slideplayer.it/slide/986973/3/images/22/%5BAla+o+Gly%5D-x-x-x-x-GlySer%5BSer+o+Thr%5D.jpg "PROSITE http :// È una banca dati che raccoglie più di 1600 motivi proteici associati ad una determinata struttura e funzione. La sintassi di PROSITE: x indica la posizione in cui ciascun residuo viene accettato. Tra le parentesi [ ] sono indicati i residui consentiti in una posizione. Tra le parentesi { } sono indicati i residui NON consentiti in una posizione. Ad esempio: [A,G]x4GK[S,T] viene tradotto come. [Ala o Gly]-x-x-x-x-GlySer[Ser o Thr] Qualche esempio pratico: Ricercare in Prosite la sequenza P68082 Scrivere l’ID dell’entry di Prosite e la famiglia a cui appartiene la proteina. Ricercare l’accession number relativo all’interleuchina 1 beta umana (usando ….) e ricercare in Prosite a quale famiglia appartiene questa proteina.")
23
Il programma ScanProsite (http://www.expasy.org/tools/scanprosite/)
confronta una sequenza con PROSITE o un motivo con tutte le sequenze proteiche riportate in SWISSPROT. …… Esempi!!

24
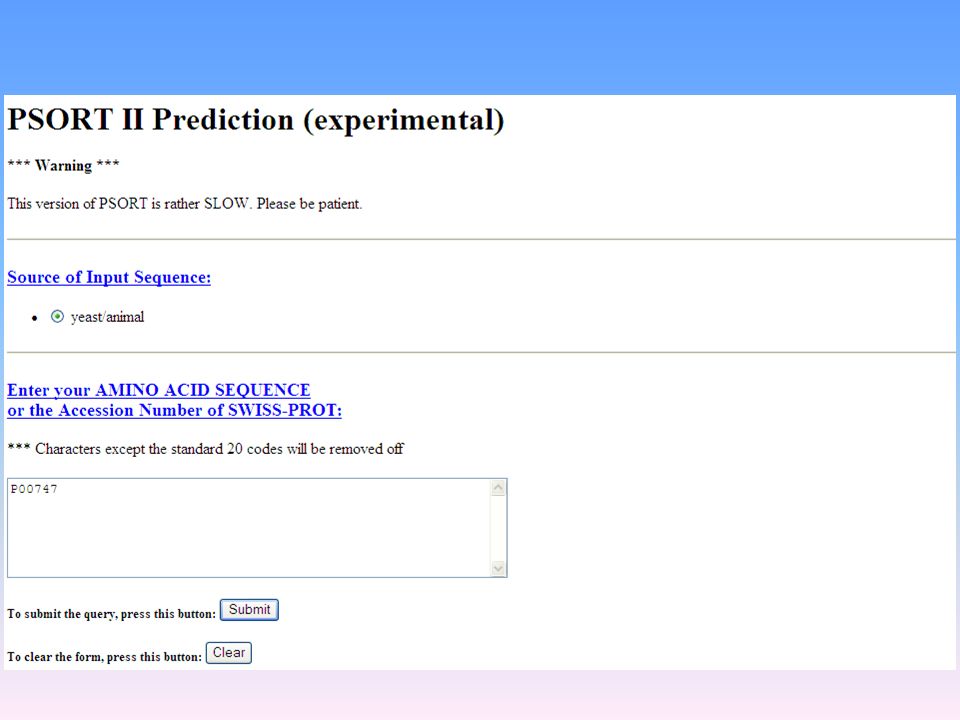
PSORT (http://psort.nibb.ac.jp/form2.html)
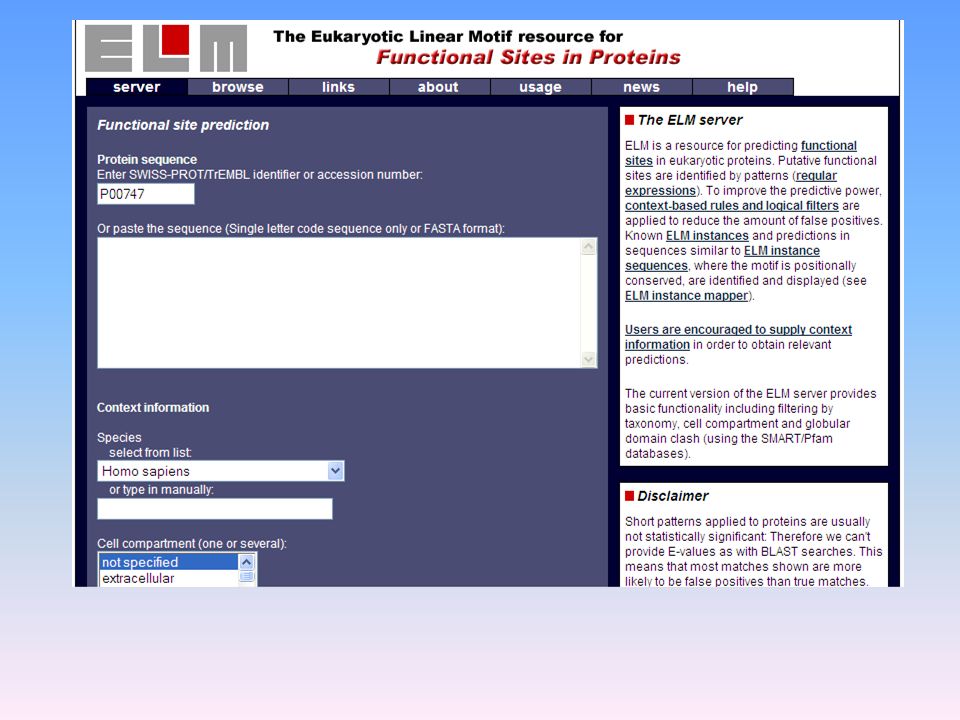
è una procedura per la predizione della localizzazione delle proteine nella cellula. Riceve informazioni sottoforma di sequenze proteiche associate a localizzazioni subcellulari e ne ricava regole di associazione empiriche. Applicando queste regole ad una sequenza proteica di localizzazione ignota, PSORT giunge a predire la localizzazione, fornendo anche un indice di affidabilità della predizione. EML ( Analizza i siti funzionali nelle proteine ESEMPI PRATICI

Presentazioni simili














 viene copiata in una sequenza complementare di RNA dall’enzima.>")




