Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
DISTRIBUZIONE DEI FARMACI

2
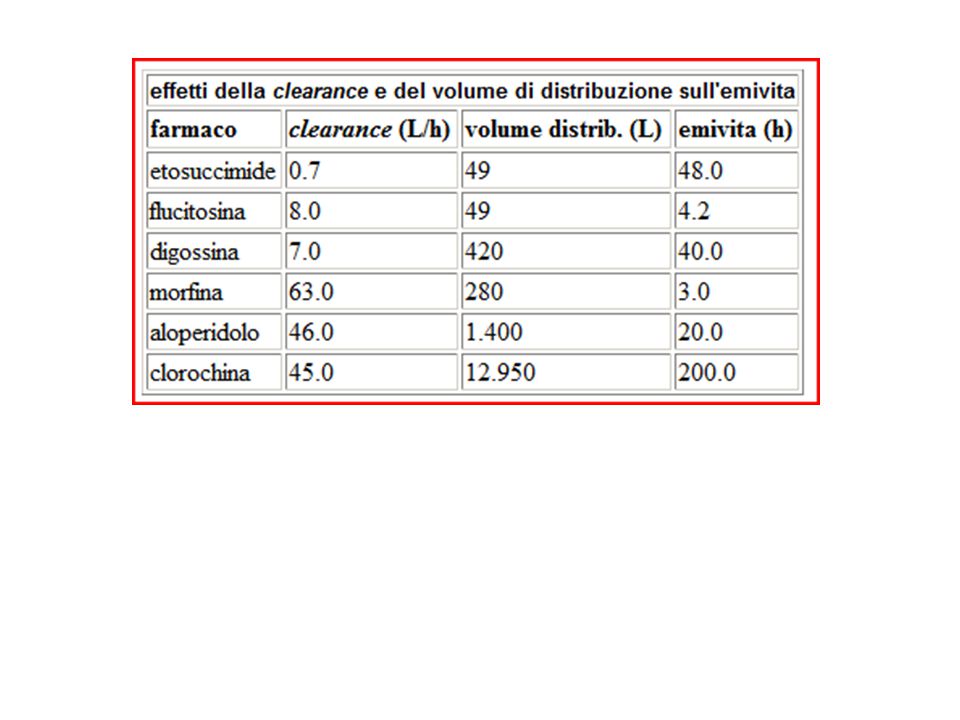
Dopo essere entrato nell’organismo, il farmaco viene distribuito ai vari tessuti ed eliminato per metabolizzazione o escrezione Bolo (150 mg) e.v. di lidocaina => la conc plasmatica decade con due velocità diverse: I -> dal sangue ai tessuti (-> equilibrio) – dipende dalla capacità del F di uscire dai vasi e dal volume in cui deve distribuirsi II -> metabolismo ed escrezione – la velocità dipende dall’efficienze dei processi di eliminazione Distribuzione ed eliminazione non sono separati nel tempo, ed è possibile distinguerli solo se uno è molto più rapido dell’altro (lidocaina). Per alcuni F, la fase di distribuzione (eq. con il compartimento extraplasmatico) può essere molto lunga e non essere riconoscibile nella curva Cp vs tempo.
 e.v. di lidocaina => la conc plasmatica decade con due velocità diverse: I -> dal sangue ai tessuti (-> equilibrio) – dipende dalla capacità del F di uscire dai vasi e dal volume in cui deve distribuirsi. II -> metabolismo ed escrezione – la velocità dipende dall’efficienze dei processi di eliminazione. Distribuzione ed eliminazione non sono separati nel tempo, ed è possibile distinguerli solo se uno è molto più rapido dell’altro (lidocaina). Per alcuni F, la fase di distribuzione (eq. con il compartimento extraplasmatico) può essere molto lunga e non essere riconoscibile nella curva Cp vs tempo.")
3
DISTRIBUZIONE: l’insieme dei processi che contribuiscono al trasferimento del farmaco ai diversi compartimenti dell’organismo. Dipende da: equilibrio: nei diversi compartimenti dell’organismo il farmaco raggiunge concentrazioni di equilibrio diverse cinetica: velocità con cui il F si distribuisce fra il sangue e i tessuti. La velocità con cui ogni tessuto di equilibra rispetto al plasma è pari, in linea di principio, al rapporto fra volume del tessuto e flusso ematico (es. tessuto adiposo scarsamente perfuso). La concentrazione del F nell’acqua interstiziale dei tessuti tenderà ad essere uguale a quella dell’acqua plasmatica. Inoltre, il F potrà legarsi a proteine e macromolecole, diffondere nelle membrane cellulari e accumularti in modo diverso nelle cellule del tessuto => la conc. del F nel tessuto sarà diversa da quella plasmatica. (per es. i farmaci lipofili si accumulano nei tessuti ricchi di lipidi)
. La concentrazione del F nell’acqua interstiziale dei tessuti tenderà ad essere uguale a quella dell’acqua plasmatica. Inoltre, il F potrà legarsi a proteine e macromolecole, diffondere nelle membrane cellulari e accumularti in modo diverso nelle cellule del tessuto => la conc. del F nel tessuto sarà diversa da quella plasmatica. (per es. i farmaci lipofili si accumulano nei tessuti ricchi di lipidi)")
4
L’avidità del tessuto per il farmaco (Kp) determina la velocità e l’entità della distribuzione
Kp = avidità di un tessuto per il farmaco (o affinità del F per un tessuto) Kp = Conc totale tissutale/Cp (all’equilibrio) Kp = Ctot tessuto Cp Una volta raggiunto l’equilibrio, Kp è costante (sia Ctot che Cp diminuiscono con l’eliminazione) finché non viene somministrata una nuova dose di farmaco. La quantità di farmaco presente in un determinato tessuto all’equilibrio dipende da: Kp (avidità tessuto-farmaco) volume del tessuto legame del farmaco alle proteine plasmatiche
 Kp = Conc totale tissutale/Cp (all’equilibrio) Kp = Ctot tessuto. Cp. Una volta raggiunto l’equilibrio, Kp è costante (sia Ctot che Cp diminuiscono con l’eliminazione) finché non viene somministrata una nuova dose di farmaco. La quantità di farmaco presente in un determinato tessuto all’equilibrio dipende da: Kp (avidità tessuto-farmaco) volume del tessuto. legame del farmaco alle proteine plasmatiche.")
5
VOLUME APPARENTE DI DISTRIBUZIONE
E’ possibile considerare l’organismo come costituito da diversi compartimenti distinti dal punto di vista funzionale.

6
VOLUME APPARENTE DI DISTRIBUZIONE
E’ possibile considerare l’organismo come costituito da diversi compartimenti distinti dal punto di vista funzionale. Tre compartimenti idrici principali: acqua plasmatica (ca. 4% del peso corporeo) liquido interstiziale (extracellulare) (ca. 16% del peso corporeo) liquidi intracellulari (ca. 40% del peso corporeo)
 liquido interstiziale (extracellulare) (ca. 16% del peso corporeo) liquidi intracellulari (ca. 40% del peso corporeo)")
7
VOLUME APPARENTE DI DISTRIBUZIONE
E’ possibile considerare l’organismo come costituito da diversi compartimenti distinti dal punto di vista funzionale. Tre compartimenti idrici principali: ADULTO MAGRO (70Kg): H2O ca 60% peso (42L) acqua plasmatica (ca. 4% del peso corporeo) - (3L) liquido interstiziale (extracellulare) (ca. 16% del peso corporeo) – (11L) liquidi intracellulari (ca. 40% del peso corporeo) - (28L)
: H2O ca 60% peso (42L) acqua plasmatica (ca. 4% del peso corporeo) - (3L) liquido interstiziale (extracellulare) (ca. 16% del peso corporeo) – (11L) liquidi intracellulari (ca. 40% del peso corporeo) - (28L)")
8
In ogni sede, una parte del F sarà anche presente (in funzione del CR) nelle membrane cellulari e nei depositi lipidici, nonché legato a proteine o ad altri accettori => la conc del F nei vari “compartimenti” sarà diversa.
 nelle membrane cellulari e nei depositi lipidici, nonché legato a proteine o ad altri accettori => la conc del F nei vari compartimenti sarà diversa.")
9
VOLUME APPARENTE DI DISTRIBUZIONE (calcolo)
Concentrazione = quantità / volume Volume in cui il F si è distribuito (eq.) = dose di F somministrata (quantità) conc plasmatica misurata (quantità/litro) Tale valore non corrisponde al volume di acqua del compartimento, bensì ad un valore apparente definito VOLUME DI DISTRIBUZIONE (Vd), che dipende dal CR, dal suo legame alle proteine plasmatiche e dall’affinità/avidità per i tessuti. Vd = Quantità di F nel corpo all’eq. di distribuzione Concentrazione plasmatica del F Vd consente di stimare la Cp all’equilibrio di distribuzione
 = dose di F somministrata (quantità) conc plasmatica misurata (quantità/litro) Tale valore non corrisponde al volume di acqua del compartimento, bensì ad un valore apparente definito VOLUME DI DISTRIBUZIONE (Vd), che dipende dal CR, dal suo legame alle proteine plasmatiche e dall’affinità/avidità per i tessuti. Vd = Quantità di F nel corpo all’eq. di distribuzione. Concentrazione plasmatica del F. Vd consente di stimare la Cp all’equilibrio di distribuzione.")
10
D = dose di farmaco somministrata
Vd = D / Cp D = dose di farmaco somministrata Cp = estrapolazione a zero della fase di eliminazione (Cp che si otterrebbe se l’intera dose somministrata raggiungesse l’equilibrio istantaneamente): Vd = (150 mg)/(2 mg/l) = 75 litri Cp = 2 mg/l NB: si preferisce esprimere il Vd come volume/Kg peso corporeo in modo da ridurre la variabilità tra individui di peso diverso (senza però tener conto di costituzione fisica, età e patologie)
: Vd = (150 mg)/(2 mg/l) = 75 litri. Cp = 2 mg/l. NB: si preferisce esprimere il Vd come volume/Kg peso corporeo in modo da ridurre la variabilità tra individui di peso diverso (senza però tener conto di costituzione fisica, età e patologie)")
11
Ogni compartimento dell’organismo contribuisce a determinare il valore complessivo di Vd:
Vd(tot) = Vplasmatico + Vd(x) + Vd(y) + Vd(z) + …… Ogni compartimento (organo o tessuto) possiede un valore di Vd: Vd(x) = Volume reale del compartimento x Kp del compartimento per il F Se Vd di un F è molto elevato (> acqua corporea) => uno o più tessuti accumulano il farmaco (Kp>1)
 = Vplasmatico + Vd(x) + Vd(y) + Vd(z) + …… Ogni compartimento (organo o tessuto) possiede un valore di Vd: Vd(x) = Volume reale del compartimento x Kp del compartimento per il F. Se Vd di un F è molto elevato (> acqua corporea) => uno o più tessuti accumulano il farmaco (Kp>1)")
13
ESEMPI DI DEPOSITO TISSUTALE:
- farmaci lipofili si accumulano nei tessuti grassi (adiposo e SNC) le tetracicline legano il calcio delle ossa i metalli pesanti si legano al tessuto osseo => danni i farmaci possono competere per i siti di deposito tissutali Il Vd e la Kp consentono di calcolare: Conc F nel tessuto (eq.) = Kp x Cp = Vd/Vreale x Cp Velocità di raggiungimento delle conc di equilibrio nel tessuto: Vel= flusso/Vd = flusso/(V x Kp) = flusso specifico/Kp
 le tetracicline legano il calcio delle ossa. i metalli pesanti si legano al tessuto osseo => danni. i farmaci possono competere per i siti di deposito tissutali. Il Vd e la Kp consentono di calcolare: Conc F nel tessuto (eq.) = Kp x Cp = Vd/Vreale x Cp. Velocità di raggiungimento delle conc di equilibrio nel tessuto: Vel= flusso/Vd = flusso/(V x Kp) = flusso specifico/Kp.")
14
IL LEGAME DEI FARMACI ALLE PROTEINE PLASMATICHE
Nel plasma: F legato alle proteine + F libero (-> distribuzione ed eliminazione) NB: nel sangue il F può anche legarsi alle cellule circolanti
 NB: nel sangue il F può anche legarsi alle cellule circolanti.")
16
ALBUMINA: proteina principale coinvolta nel legame di sostanze endogene e esogene costituisce circa il 50% delle proteine plasmatiche presenta circa 100 gruppi carichi al pH fisiologico vi si legano principalmente gli ANIONI organici (es. acidi grassi liberi, bilirubina, penicillina, sulfonamidi, salicilati, barbiturici, acido ascorbico). a1 GLICOPROTEINA ACIDA: importante per il trasporto di piccole molecole a1 immunoglobulina con un sito ad alta affinità per molecole basiche TRANSCORTINA: a1-proteina legante gli estrogeni PROTEINE DEI GRUPPI a2, b1, b2: - trasportano lipidi, colesterolo, vitamine liposolubili, emoglobina (aptoglobina), ioni zinco, rame (ceruloplasmina) e ferro (transferrina).
. a1 GLICOPROTEINA ACIDA: importante per il trasporto di piccole molecole. a1 immunoglobulina con un sito ad alta affinità per molecole basiche. TRANSCORTINA: a1-proteina legante gli estrogeni. PROTEINE DEI GRUPPI a2, b1, b2: - trasportano lipidi, colesterolo, vitamine liposolubili, emoglobina (aptoglobina), ioni zinco, rame (ceruloplasmina) e ferro (transferrina).")
17
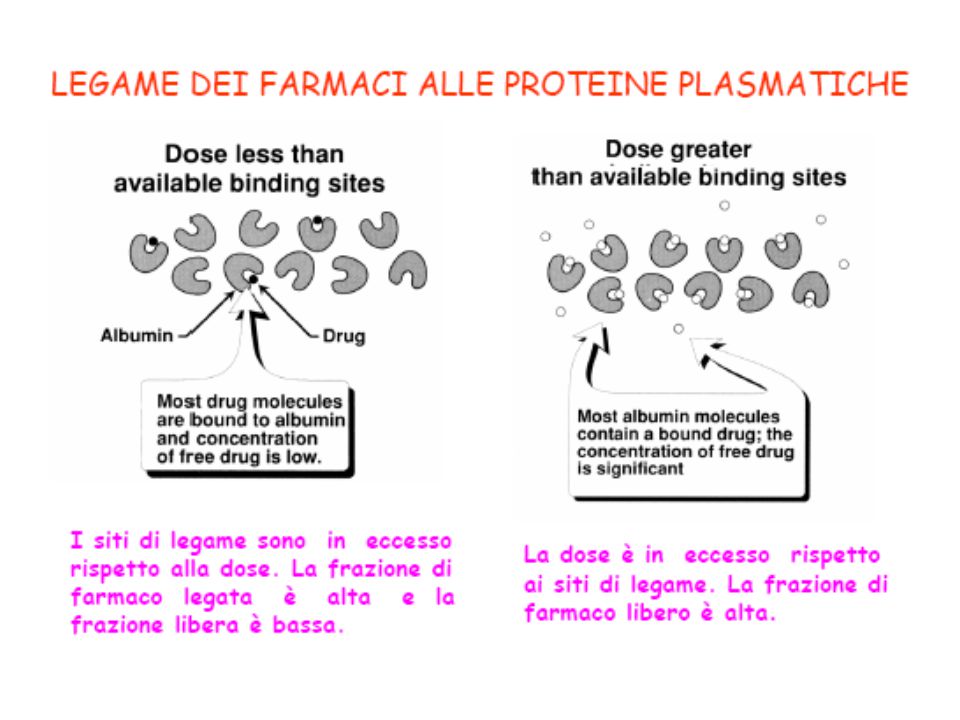
SPIAZZAMENTO DAL LEGAME CON LE PROTEINE PLASMATICHE
Il legame alle proteine plasmatiche è di solito non selettivo => COMPETIZIONE E SPIAZZAMENTO Lo spiazzamento è importante per farmaci con basso IT, fortemente legati alle proteine plasmatiche (>90%) e con Vd piuttosto limitato [possibili effetti indesiderati o tossici] I farmaci acidi (salicilati, fenilbutazione, sulfamidici) possono spiazzare: il warfarin (anticoagulante orale, F-P 98%) => gravi episodi emorragici la tolbutamide (ipoglicemizzante orale) => crisi ipoglicemiche la fenitoina (antiepilettico) I sulfamidici spiazzano la bilirubina => complicazioni iatrogene dell’ittero postnatale (la bilirubina si deposita nei gangli della base e nei nuclei subtalamici => encefalopatia tossica: kernittero). Epatopatie => ipoalbuminemia => aumento conc F libero
 e con Vd piuttosto limitato [possibili effetti indesiderati o tossici] I farmaci acidi (salicilati, fenilbutazione, sulfamidici) possono spiazzare: il warfarin (anticoagulante orale, F-P 98%) => gravi episodi emorragici. la tolbutamide (ipoglicemizzante orale) => crisi ipoglicemiche. la fenitoina (antiepilettico) I sulfamidici spiazzano la bilirubina => complicazioni iatrogene dell’ittero postnatale (la bilirubina si deposita nei gangli della base e nei nuclei subtalamici => encefalopatia tossica: kernittero). Epatopatie => ipoalbuminemia => aumento conc F libero.")
18
Interazioni a livello del legame farmaco-proteico
Farmaci che si legano tenacemente alle proteine plasmatiche: FANS (fenilbutazone, ac. acetilsalicilico), difenilidantoina, fenotiazine. I FANS spiazzano: gli ipoglicemizzanti orali dalle proteine plasmatiche (↓ glicemia coma ipoglicemico) gli anticoagulanti (es: warfarin) dalle proteine plasmatiche (rischio di emorragie gravi)
, difenilidantoina, fenotiazine. I FANS spiazzano: gli ipoglicemizzanti orali dalle proteine plasmatiche (↓ glicemia coma ipoglicemico) gli anticoagulanti (es: warfarin) dalle proteine plasmatiche (rischio di emorragie gravi)")
19
FATTORI CHE INFLUENZANO LA VELOCITA’ DI DISTRIBUZIONE
1) Permeabilità locale (CR, presenza di barriere, etc etc) 2) Perfusione ematica (farmaci lipofili si accumulano rapidamente nei tessuti più perfusi) 3) Volume del tessuto (> V, > tempo per raggiungere l’equilibrio sangue-tessuto) 4) Avidità del tessuto per il farmaco (>Kp, > tempo per raggiungere l’equilibrio s-t) t1/2 = 0,693 x Kp x V flusso t1/2 = emivita di distribuzione: tempo necessario perché la differenza di conc tra i due compartimenti si dimezzi
 Permeabilità locale (CR, presenza di barriere, etc etc) 2) Perfusione ematica (farmaci lipofili si accumulano rapidamente nei tessuti più perfusi) 3) Volume del tessuto (> V, > tempo per raggiungere l’equilibrio sangue-tessuto) 4) Avidità del tessuto per il farmaco (>Kp, > tempo per raggiungere l’equilibrio s-t) t1/2 = 0,693 x Kp x V. flusso. t1/2 = emivita di distribuzione: tempo necessario perché la differenza di conc tra i due compartimenti. si dimezzi.")
20
L’avidità del tessuto per il farmaco (Kp) determina la velocità e l’entità della distribuzione
 determina la velocità e l’entità della distribuzione")
21
Il flusso ematico di ciascun compartimento determina la velocità e l’entità della distribuzione
Il tessuto adiposo, scarsamente perfuso, può non riuscire ad equilibrarsi con il plasma prima che il F venga eliminato o seguire le conc plasmatiche con notevole ritardo

Presentazioni simili