Scaricare la presentazione
La presentazione è in caricamento. Aspetta per favore

1
ISMETT, Palermo, 23.03.2012 Enrico Geraci Problemi etici nella Ricerca Clinica

2
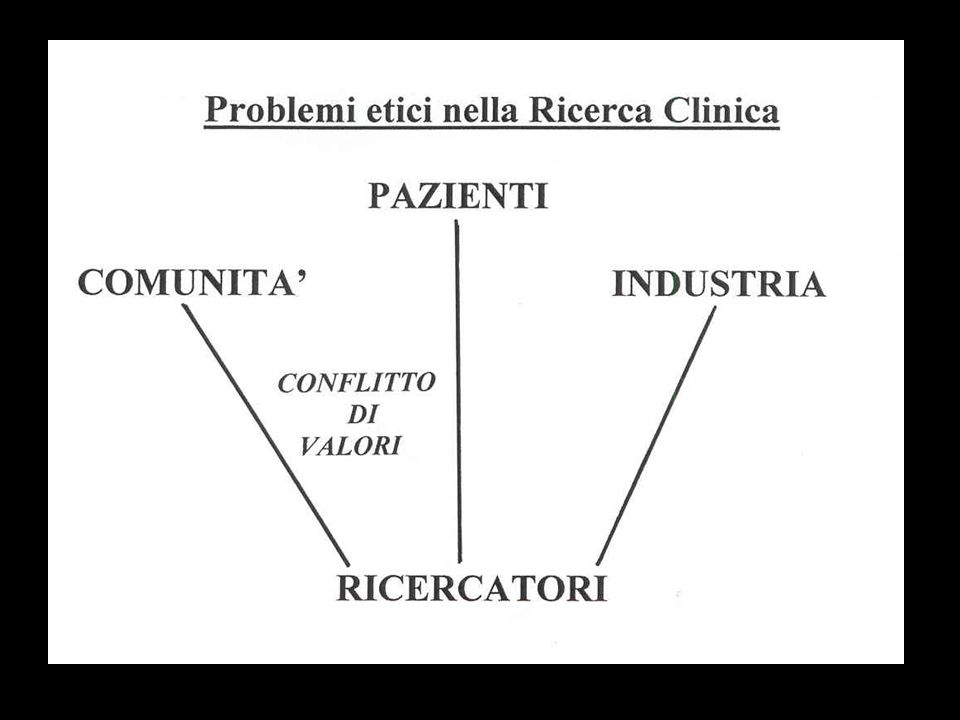
La maggior parte dei problemi etici nella ricerca clinica si configura nello ambito dei rapporti fra Medici Ricer- catori, Pazienti arruolati nella ricerca, Comunità (comprendente gli altri pa zienti, presenti e futuri) e Industria (dei farmaci, dei devices) che spesso promuove e molto spesso finanzia la ricerca clinica.
 e Industria (dei farmaci, dei devices) che spesso promuove e molto spesso finanzia la ricerca clinica.")
3
Schematicamente tali rapporti si possono raffigurare ponendo al centro i Ricercatori…

5
Possono emergere per i Ricercatori problemi etici di scelta fra il bene dei Pazienti arruolati nella ricerca e il be ne della Comunità. In questi casi pre ferisco parlare di conflitto di valori (ne vedremo un esempio più in là)…
….")
7
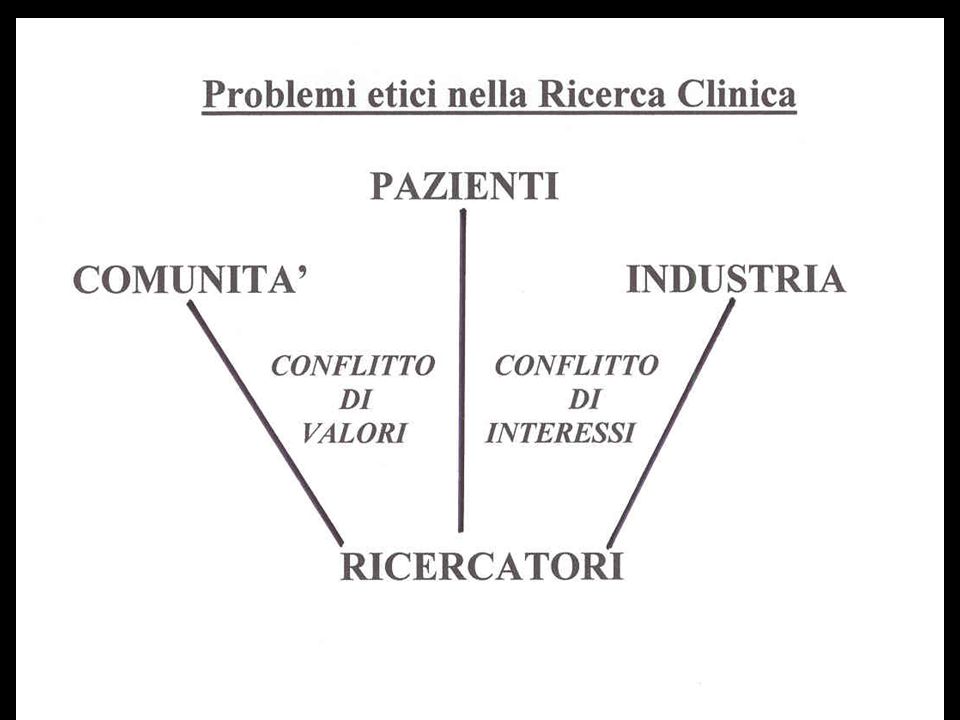
Ma più frequentemente entrano in competizione il bene dei Pazien ti e quello dell’ Industria, oppure il bene dei Pazienti e quello dei Ri cercatori, e allora si deve parlare di conflitto di interessi…

9
- Il conflitto di valori ha sempre implicazioni etiche, più o meno proble matiche. - Il conflitto di interessi può costi tuire un problema etico, ma talora im plica piuttosto problemi di correttezza professionale (ricercatori) o commercia le (industria), e in qualche caso si può sconfinare in comportamenti illeciti, configuranti ‘solo’ una frode scientifi
 o commercia le (industria), e in qualche caso si può sconfinare in comportamenti illeciti, configuranti ‘solo’ una frode scientifi.")
10
ca o persino un vero e proprio reato. Mentre è chiaro a tutti che l’industria persegue prioritariamente il profitto, sì che si tende a guardare con grande at tenzione ai connessi potenziali conflitti d’interessi, non sempre si tiene adegua to conto del fatto che i ricercatori han no interessi di carriera e di prestigio, e che possono anche essere tentati da be nefici economici …

11
Ma non sempre l’esistenza di un con flitto di interessi in questo contesto rende impossibile la partecipazione ad un trial clinico come ricercatore. Dipende dalla natura e dall’ intensità del conflitto. Ad esempio, è sicuro im pedimento un forte legame con l’ in dustria finanziatrice (possesso di azio ni, etc), mentre è accettato che il can didato ricercatore abbia tenuto confe renze rimunerate da quell’ industria.
, mentre è accettato che il can didato ricercatore abbia tenuto confe renze rimunerate da quell’ industria..")
12
Anni sessanta del 1900 : due interventi fondamentali sulla ricerca clinica non-etica :

13
SPECIAL ARTICLE ------------- ETHICS AND CLINICAL RESEARCH HENRY K. BEECHER, M.D. NEJM 1966; 274: 1354-1360

14
Commentava 22 esempi di ricerca clini ca non-etica, con particolare attenzione ai problemi del ‘consenso informato’. “Abitualmente i pazienti non intendo no rischiare consapevolmente la loro sa lute o la loro vita per il bene della ‘scien za’. Ogni esperto sperimentatore lo sa. Quando tali rischi vengono corsi ed è co involto un numero considerevole di pazi enti, si deve assumere che il consenso in formato non è stato ottenuto da tutti.”

15
M. H. Pappworth Human Guinea Pigs – here and now Experimentation on man Routledge & Kegan Ltd, London, 1967

16
“Lo sperimentatore non deve conside rare, proporre o intraprendere alcun esperimento [sull’uomo] ove questo sia tale che - in circostanze identiche a quelle riguardanti il paziente - lo spe rimentatore esiterebbe a sottoporvi sè stesso, o membri della sua famiglia o al tre persone per le quali egli senta rispet to e affetto.”
![Lo sperimentatore non deve conside rare, proporre o intraprendere alcun esperimento [sull’uomo] ove questo sia tale che - in circostanze identiche a quelle riguardanti il paziente - lo spe rimentatore esiterebbe a sottoporvi sè stesso, o membri della sua famiglia o al tre persone per le quali egli senta rispet to e affetto.](http://images.slideplayer.it/10/2808546/slides/slide_16.jpg "Lo sperimentatore non deve conside rare, proporre o intraprendere alcun esperimento [sull’uomo] ove questo sia tale che - in circostanze identiche a quelle riguardanti il paziente - lo spe rimentatore esiterebbe a sottoporvi sè stesso, o membri della sua famiglia o al tre persone per le quali egli senta rispet to e affetto.")
17
Giulio Maccacaro (dalla Prefazione alla edizione italiana del Pappworth, 1971) : A. Sperimentazione nell’ uomo : Necessaria (ma tipo B. o C. ?) B. Sperimentazione sull’ uomo : Abituale ma da condannare. C. Sperimentazione con l’ uomo : Auspicabile ma molto difficile.
 B. Sperimentazione sull’ uomo : Abituale ma da condannare. C. Sperimentazione con l’ uomo : Auspicabile ma molto difficile..")
18
In seguito, i protagoni sti della vicenda sono di venuti i Comitati Etici…

19
I Comitati Etici per la Ricerca Clinica (con particolare riferimento al trial clinico randomizzato (RCT, da Randomized Clinical Trial)
")
20
I Comitati Etici per la Ricerca Clinica hanno come compito fondamentale la protezione dei pazienti arruolati negli studi. Sono ovviamente chiamati a pro nunciarsi sugli eventuali conflitti di valori (Pazienti / Comunità), ma buona parte del loro impegno consiste nel pro teggere sia i pazienti che l’ integrità del la ricerca dai conflitti di interessi (Ri cercatori / Pazienti / Industria).
, ma buona parte del loro impegno consiste nel pro teggere sia i pazienti che l’ integrità del la ricerca dai conflitti di interessi (Ri cercatori / Pazienti / Industria)..")
21
Comitati etici per i Trial Clinici Randomizzati

22
1) Comitato Etico interno al trial, chia mato da qualche tempo Data and Safe ty Monitoring Board (DSMB). 2) Comitati Etici esterni al trial, di singoli Ospedali, di gruppi di Ospeda li, di Policlinici, o anche a livello centra le regionale, detti in Italia di solito Co mitati Etici Locali ma in campo inter nazionale chiamati Institutional Revi ew Boards (IRBs).
 Comitati Etici esterni al trial, di singoli Ospedali, di gruppi di Ospeda li, di Policlinici, o anche a livello centra le regionale, detti in Italia di solito Co mitati Etici Locali ma in campo inter nazionale chiamati Institutional Revi ew Boards (IRBs)..")
23
Comitato etico interno al trial DSMB (Data and Safety Monitoring Board)
")
24
Il DSMB è il comitato etico indipen dente interno al trial. Si occupa solo di quel dato trial, collaborando con lo Ste ering Committee prima, durante e an che dopo la fine dello studio. Il DSMB esamina il protocollo del tri al prima che lo studio inizi e può richie dere modifiche prima di approvarlo, poi garantisce il corretto svolgimento della ricerca, e infine controlla l’appro

25
priatezza dell’ elaborazione dei dati e della presentazione dei risultati. I DSMB vennero introdotti nella ri cerca clinica negli anni ‘60 del 1900 dall’NIH (USA) per monitorare i dati delle analisi ad interim* nei trial clini ci onde proteggere i pazienti arruolati. *risultati parziali raccolti in un dato mo mento (abitualmente prestabilito) duran te lo svolgimento dello studio.
 per monitorare i dati delle analisi ad interim* nei trial clini ci onde proteggere i pazienti arruolati. *risultati parziali raccolti in un dato mo mento (abitualmente prestabilito) duran te lo svolgimento dello studio..")
26
In seguito i compiti del DSMB si sono estesi agli altri aspetti etici e a quelli me todologici del trial.Tuttavia la sicurezza dei pazienti rimane il compito principale del DSMB, che a tal fine si tiene informa to, nel corso dello studio, sui risultati di eventuali analoghe ricerche altrui che via via vengano resi noti, e valuta in mo do esclusivo e indipendente i risultati del le analisi ad interim del proprio trial…

27
In base a tali informazioni / valuta zioni il DSMB può raccomandare : a) che lo studio prosegua invariato; b) che venga modificato in itinere il Protocollo; c) che il trial venga chiuso in anticipo.
 che lo studio prosegua invariato; b) che venga modificato in itinere il Protocollo; c) che il trial venga chiuso in anticipo.")
28
Quando è necessario un DSMB # Quando un trial potrebbe rivelare dif ferenze significative in tangible human outcomes (concrete [gravi] conseguen ze per l’ uomo) : ad esempio, nei trial cardiologici, quando gli end points sono la morte, l’ infarto, l’ ictus. # Quando uno dei trattamenti da testa re nel trial comporta notoriamente pos sibili effetti collaterali gravi.
![Quando è necessario un DSMB # Quando un trial potrebbe rivelare dif ferenze significative in tangible human outcomes (concrete [gravi] conseguen ze per l’ uomo) : ad esempio, nei trial cardiologici, quando gli end points sono la morte, l’ infarto, l’ ictus.](http://images.slideplayer.it/10/2808546/slides/slide_28.jpg "# Quando uno dei trattamenti da testa re nel trial comporta notoriamente pos sibili effetti collaterali gravi..")
29
# Quando il rischio dei trattamenti da testare nel trial non è ben conosciuto (ad esempio, negli studi di terapia geni ca) e potrebbe essere alto. # Quando un trial valuta una terapia già nota, ma per un nuovo impiego, specie se si sa che tale terapia compor ta un significativo profilo di rischio. Ma un DSMB può essere comun que istituito anche in altri casi…

31
Circa 1800 pazienti avviati a CABG elettivo, vennero randomizzati ad uno dei tre seguenti gruppi : 1) Pazienti che ricevettero (senza sa perlo) la preghiera di suffragio, dopo essere stati informati che avrebbero po tuto riceverla o non riceverla. 2) Pazienti che non ricevettero (sen za saperlo) la preghiera, dopo essere stati informati che avrebbero potuto riceverla o non riceverla.
 Pazienti che non ricevettero (sen za saperlo) la preghiera, dopo essere stati informati che avrebbero potuto riceverla o non riceverla..")
32
3) Pazienti che ricevettero la preghiera dopo essere stati infor mati che l’avrebbero ricevuta. Primary end point : any compli cation within 30 days of CABG.

34
Risultati del trial STEP pubblicati nel l’ aprile 2006 (Benson H et al, Am Heart J 2006; 151: 934 -942) : “Conclusions : Intercessory prayer itself had no effect on complication- free recovery from CABG, but certain ty of receiving the intercessory prayer was associated with a higher incidence of complications.”
 : Conclusions : Intercessory prayer itself had no effect on complication- free recovery from CABG, but certain ty of receiving the intercessory prayer was associated with a higher incidence of complications.")
35
Composizione del DSMB A questo proposito non vi è accordo unanime nella comunità della ricerca clinica, se non sul fatto che il numero dei componenti deve essere limitato. Un DSMB con più di 5 o 6 membri rischia di lavorare male (difficoltà di riunirsi, problemi nelle comunicazioni tra i membri, etc.)…
….")
36
Di certo devono far parte del DSMB clinici esperti nel campo concernente quel dato trial ma anche competenti in metodologia della ricerca clinica, nonché uno statistico. Si discute sulla presenza di un esperto in bioetica e di un laico rappresentante dei pazienti arruolandi nel trial. Comunque il DSMB può avvalersi, quando lo giudica opportuno, di speci fici consulenti temporanei.

37
Il DSMB è rigorosamente indipenden te. I suoi membri non devono avere for ti legami (di carriera, economici, etc) con i ricercatori o con i finanziatori del trial. La sorveglianza del DSMB su etici tà e correttezza metodologica del trial si esercita: 1.Nelle fasi preliminari dello studio. 2.Durante lo svolgimento del trial. 3.Dopo la conclusione dello studio.
 con i ricercatori o con i finanziatori del trial. La sorveglianza del DSMB su etici tà e correttezza metodologica del trial si esercita: 1.Nelle fasi preliminari dello studio. 2.Durante lo svolgimento del trial. 3.Dopo la conclusione dello studio..")
38
Compiti del DSMB nelle fasi preliminari dello studio

39
# Verifica della equipoise di parten za, della potenziale utilità del progetto (per i pazienti arruolandi e/o per i pa zienti futuri) e della sua praticabilità, nonché dell’ assenza di incompatibili tà culturali, religiose, etniche. # Verifica dell’ adeguatezza metodolo gica del Protocollo del trial.

40
# Verifica dell’assenza di inaccet tabili conflitti di interessi nei ricer catori. # Verifica della correttezza della procedura per il Consenso Infor mato.

41
Equipoise Per ovvi motivi etici è necessario che i ricercatori di un trial clinico randomiz zato siano, in partenza, in una condizio ne di incertezza sulla rispettiva effica cia dei trattamenti che verranno con frontati nel trial. Nel mondo della ricer ca clinica tale condizione è chiamata equipoise (bilanciamento, equilibrio)…
….")
42
Ma non è necessario che ogni singolo ricercatore sia ‘in equipoise’ prima che il trial inizi, purchè esista nella comuni- tà clinica una diffusa incertezza sul ri spettivo valore dei trattamenti che si vo gliono testare : si parla allora di clini cal equipoise.

43
Ovviamente le cose non sempre sono semplici : si veda il caso del trial COU RAGE (2007), che dimostrò che, nella cardiopatia ischemica stabile, la tera pia solo medica non è inferiore agli in terventi coronarici percutanei (PCI : angioplastica, stent, etc). Il trial venne iniziato tardi perché molti cardiologi interventisti lo giudicavano non-etico (insufficiente equipoise).
..")
44
Consenso informato nella Ricerca Clinica

45
_____________________________ Approaches to Informed Consent Gianni Tognoni, MD and Enrico Geraci, MD Controlled Clin Trials 1997; 18: 621-627

46
Ai pazienti da arruolare vanno forni te esaurienti informazioni su : scopo della ricerca, procedure, potenziali ri schi e benefìci, possibili alternative. E’ importante fare in modo che il paziente comprenda bene le informazioni fonite gli, onde possa decidere davvero libera mente se partecipare allo studio…

47
Ma vi sono situazioni nelle quali non è possibile ottenere un valido consenso direttamente dal paziente, e si deve ri correrre ad un surrogato : ai genitori nei trial condotti nei bambini, a fami liari nel caso di persone mentalmente incapaci, etc.

48
Una situazione di particolare difficol tà è quella dei trial clinici randomizzati che si intendano condurre in pazienti in condizioni di emergenza, come ad es. nell’ infarto miocardico acuto in fase iniziale. Sottoporre tali pazienti ad una corretta procedura per ottenerne il con senso comporterebbe per loro due tipi di danno :

49
1) In una persona già molto preoccu pata, ma inizialmente non pienamen te informata, l’apprendere di avere un infarto provocherebbe quasi certamen te ulteriore grave ansia o angoscia. 2) La procedura per il consenso, se è eseguita seriamente, consuma tempo e comporterebbe un nocivo ritardo nel ricevere la terapia potenzialmente sal vavita (trombolisi o PCI primario).
 La procedura per il consenso, se è eseguita seriamente, consuma tempo e comporterebbe un nocivo ritardo nel ricevere la terapia potenzialmente sal vavita (trombolisi o PCI primario)..")
50
Nel primo trial GISSI (1986), che ha onorato nel mondo la Cardiologia Ita liana (prima dimostrazione dell’effica cia della trombolisi precoce nell’ infar to miocardico acuto), lo Steering Com mittee, presieduto da Fausto Rovelli (Co-chair Gianni Tognoni) si assunse formalmente la responsabilità di fare a meno del consenso informato dei pa zienti, per non danneggiarli…
, che ha onorato nel mondo la Cardiologia Ita liana (prima dimostrazione dell’effica cia della trombolisi precoce nell’ infar to miocardico acuto), lo Steering Com mittee, presieduto da Fausto Rovelli (Co-chair Gianni Tognoni) si assunse formalmente la responsabilità di fare a meno del consenso informato dei pa zienti, per non danneggiarli…")
51
Altrimenti, per rispettare l’ autono mia dei pazienti (principio etico cui fa capo il ‘consenso informato’), si sareb be dovuto rinunciare ad eseguire uno studio che si auspicava potesse arreca re, se positivo, un grande beneficio al la Comunità. Si era in una chiara condizione di con flitto di valori (Pazienti vs Comunità) risolto in favore della Comunità…
 risolto in favore della Comunità….")
52
Vi furono critiche, che però si spense ro quando fu evidente l’ importanza del beneficio per la Comunità derivato dal GISSI (in seguito chiamato GISSI-1). Pochi mesi fa vi è stato su questo tema un intervento autorevole e deciso :

53
Effect of consent rituals on mortality in emergency care research (Letter) Ian Roberts, David Prieto-Merino, Halee ma Shakur, Iain Chalmers, John Nicholl Clinical Trials Unit, London School of Hy giene and Tropical Medicine Lancet 2011; 377: 1071-2
 Ian Roberts, David Prieto-Merino, Halee ma Shakur, Iain Chalmers, John Nicholl Clinical Trials Unit, London School of Hy giene and Tropical Medicine Lancet 2011; 377:")
54
“We argue that the need for an urgent trial treatment, even in patients who are conscious and whose relatives are available, by itself excludes the possibi lity of fully informed consent. If consent rituals delay the start of a trial treatment such that the treatment effect could be reduced or obscured, we maintain that seeking consent is ac tually unethical.”

55
A proposito di consenso nella ricerca, è interessante prendere atto di una proposta avanzata pochi anni fa da alcuni eminen ti eticisti (non mi risulta finora in odore di realizzazione…) :
 :")
56
The Obligation to Participate in Biomedical Research GO Schaefer, EJ Emanuel, A Wertheimer JAMA, July 1, 2009

57
“L’ opinione che attualmente pre vale è che la partecipazione alla ricer ca biomedica non sia un preciso dove re. Ma alcuni commentatori hanno avanzato motivi contrari a questa po sizione, e noi proponiamo adesso un nuovo argomento ‘di bene pubblico’ per un obbligo a partecipare [da sog getto / paziente] a tale ricerca…

58
La ricerca biomedica è infatti un bene pubblico, [i cui risultati sono] a disposizione di ogni individuo an che se egli non vi ha partecipato… La partecipazione alla ricerca bio medica è un mezzo di rilevanza cri tica per supportare un importante bene pubblico. Di conseguenza, tut ti hanno l’obbligo di parteciparvi…
![La ricerca biomedica è infatti un bene pubblico, [i cui risultati sono] a disposizione di ogni individuo an che se egli non vi ha partecipato… La partecipazione alla ricerca bio medica è un mezzo di rilevanza cri tica per supportare un importante bene pubblico.](http://images.slideplayer.it/10/2808546/slides/slide_58.jpg "Di conseguenza, tut ti hanno l’obbligo di parteciparvi….")
59
La norma sociale corrente è che gli individui partecipano solo se essi han no buone ragioni per farlo. L’ argo mento ‘bene pubblico’ implica invece che gli individui debbano partecipare alla ricerca biomedica [come sogget ti / pazienti] tranne nel caso in cui ab biano un buon motivo per non farlo.

60
Questo cambio di posizione dovrebbe essere di grande aiuto al progresso della ricerca biomedica, con la prospettiva di rendere la società significativamente più in salute e longeva.” Department of Bioethics, The Clinical Center, National Institutes of Health Bethesda, Maryland “Questo articolo chiede un cambiamen to culturale e morale, non uno legale”

61
PAUSA

62
Compiti del DSMB nel corso dello studio

63
Per proteggere i pazienti arruolati il DSMB deve prestare grande attenzione ad eventuali variazioni, interne o ester ne al trial, che possano venire a modifi- care in misura critica la condizione di equipoise di partenza. Infatti in tal caso, se il trial continuasse come programma to, i pazienti di un braccio non fruireb bero del miglior trattamento adesso ac certato…

64
Chiusura precoce di un RTC L’ eventuale percezione di importanti variazioni dell’ equipoise mentre lo stu dio è in corso (ad es. se una analisi ad interim mostra la netta superiorità di uno dei trattamenti) deve imporre al DSMB di prendere in seria considera zione la chiusura anticipata del trial…
 deve imporre al DSMB di prendere in seria considera zione la chiusura anticipata del trial….")
65
Va ricordato che all’ epoca della lo ro istituzione il compito dei DSMB e ra, appunto, solo quello di proporre, quando opportuno, l’ interruzione pre coce del trial in base ai risultati delle proprie analisi ad interim. N.B. Attualmente si può interrompere un trial anche nel caso che ripetute ana lisi ad interim dimostrino che non vi sa ranno vincitori (chiusura per ‘futilità’).
..")
66
Ma l a chiusura anticipata di un RCT perchè uno dei trattamenti si dimostra già in itinere chiaramente superiore, ri chiede delicate valutazioni etiche, stati stiche e pratiche. Pertanto il DSMB non si deve basare solo sull’ analisi ad interim dei dati del proprio trial, ma de ve tenere anche conto :

67
a) Dell’ intero contesto del trial : enti tà dei potenziali vantaggi o rischi per i pazienti; rilevanza dei risultati del trial per la pratica clinica generale; aspetti organizzativi, pratici, economi ci. b) Delle notizie che giungono dallo esterno : risultati di altri RCT analo ghi che intanto si sono conclusi (che po trebbero mostrare chiari vantaggi o
 Delle notizie che giungono dallo esterno : risultati di altri RCT analo ghi che intanto si sono conclusi (che po trebbero mostrare chiari vantaggi o.")
68
svantaggi del trattamento sperimenta le) ; risultati di metaanalisi di prece denti studi analoghi ; eventuale eviden ziazione di effetti collaterali inaccetta bili del nuovo farmaco studiato ; etc. Si noti che i dati ‘esterni’, se da una parte possono contribuire alla chiusu ra anticipata di un trial, dall’ altra do vrebbero rendere il DSMB guardin go sulla validità della propria analisi

69
ad interim nei casi in cui essa indur rebbe alla chiusura precoce del trial mentre i dati esterni non depongono in tal senso. In taluni casi la risposta del DSMB ai risultati dell’ analisi ad interim e/o dei dati ‘esterni’ può consistere non nella chiusura anticipata del trial ma nella modifica in itinere del Protocol- lo dello studio. (In tal caso va rinno vato il ‘consenso informato’)…
….")
70
Un esempio è rappresentato da quan to accadde nel trial GISSI-Prevenzio ne, svoltosi in Italia negli anni novan ta del 1900 e noto soprattutto per aver mostrato che gli acidi grassi poli insaturi omega-3 riducono il rischio di morte improvvisa nel post-infarto negli infartuati a rischio globale me dio o basso (come erano i pazienti ar ruolati in quel trial)…
…")
71
Ma il GISSI-Prevenzione studiava an che la pravastatina contro placebo nei pazienti post-infarto. E vi furono deli cati problemi decisionali quando, piut tosto presto nel corso del trial (iniziato nel 1993), furono disponibili i risultati di due altri grandi studi in parte simi li : dapprima quelli del 4S (1994), con dotto con simvastatina, poi…
, furono disponibili i risultati di due altri grandi studi in parte simi li : dapprima quelli del 4S (1994), con dotto con simvastatina, poi….")
72
quelli del CARE (1996), con pravasta tina. Entrambi mostravano chiara mente che le statine riducono morbi lità e mortalità in pazienti con cardio patia ischemica o in individui ad alto rischio di contrarre tale patologia… Io facevo parte dello Steering Commit tee del GISSI-Prevenzione, e Luigi Pa gliaro era il Chairman del DSMB…

73
[Quando arrivarono i dati del 4S e del CARE non era ancora disponibile la prima analisi ad interim del GISSI- Prevenzione] DSMB e Steering Committee decisero dapprima, in risposta al 4S, di modifi care il Protocollo escludendo i pazien ti con colesterolemia > 250 mg/dl. Ma dopo il CARE il ramo pravastatina venne chiuso definitivamente.
![[Quando arrivarono i dati del 4S e del CARE non era ancora disponibile la prima analisi ad interim del GISSI- Prevenzione] DSMB e Steering Committee decisero dapprima, in risposta al 4S, di modifi care il Protocollo escludendo i pazien ti con colesterolemia > 250 mg/dl.](http://images.slideplayer.it/10/2808546/slides/slide_73.jpg "Ma dopo il CARE il ramo pravastatina venne chiuso definitivamente..")
74
Indebite ingerenze dell’industria: a) Pressione per interrompere precoce- mente il trial : 1. Perché l’industria non ritiene più che il suo farmaco possa vin cere (“soldi sprecati”). 2. Perché nella analisi ad interim il nuovo farmaco sem bra vincente e l’ industria non vuole cor rere il rischio che proseguendo il trial la superiorità non venga confermata.
. 2. Perché nella analisi ad interim il nuovo farmaco sem bra vincente e l’ industria non vuole cor rere il rischio che proseguendo il trial la superiorità non venga confermata..")
75
b) Pressione per non interrompere il trial benchè sia diventato evidente che il nuovo trattamento è superiore (per analisi ad interim e/o per l’ arrivo di dati esterni), onde trarre il massimo vantaggio promozionale…
 Pressione per non interrompere il trial benchè sia diventato evidente che il nuovo trattamento è superiore (per analisi ad interim e/o per l’ arrivo di dati esterni), onde trarre il massimo vantaggio promozionale…")
76
Molto grave fu il caso, negli anni ’80 - ’90 del 1900, dei trial sugli allora nuovi e costosi trombolitici (rt-PA, APSAC) nell’ infarto miocardico acuto, trial por tati avanti (o addirittura iniziati) contro placebo quando era già evidente, grazie ad analoghi trial eseguiti con la ben me no costosa streptochinasi, che la trombo lisi tempestiva nell’infarto acuto è salva vita…
 nell’ infarto miocardico acuto, trial por tati avanti (o addirittura iniziati) contro placebo quando era già evidente, grazie ad analoghi trial eseguiti con la ben me no costosa streptochinasi, che la trombo lisi tempestiva nell’infarto acuto è salva vita…")
77
A metà dei pazienti con infarto acuto arruolati in tali trial (i pazienti del brac cio placebo) venne dunque negata una terapia salvavita di ormai provata effi cacia, la trombolisi, onde poter dimo strare facilmente anche la validità dei nuovi trombolitici molto più costosi del la streptochinasi…
 venne dunque negata una terapia salvavita di ormai provata effi cacia, la trombolisi, onde poter dimo strare facilmente anche la validità dei nuovi trombolitici molto più costosi del la streptochinasi…")
78
I risultati di questi trial gravemen te non-etici vennero tutti pubblicati nelle maggiori riviste mediche, e in tutti i casi veniva dichiarato che era stato ottenuto dai pazienti il ‘consen so informato’! E con ben poche cri tiche.Una mia lettera di energica pro testa, pubblicata sul JACC, non eb be grande eco…

79
Enrico Geraci (letter): Unethical placebo assignment in clinical trials of thrombolysis. J Am Coll Cardiol 1992; 20: 1902-3

80
Compiti del DSMB alla fine del trial

81
Sorveglianza su : # Sc orretto assemblaggio e inappropria ta valutazione dei risultati dello studio. # Sc orrette modalità di presentazione dei risultati. # Ritardo nella pubblicazione, o rinun cia alla pubblicaz., di risultati negativi. # Diffusione precoce di risultati favore voli, senza il vaglio di peer reviewers.

82
Spesso vi è incertezza su come elabo- rare e presentare i risultati di un trial : Analisi intention-to-treat o per-proto col ? Riduzione relativa o assoluta de gli end points ? Etc. Tuttavia tali dubbi non sempre sono genuini, e la scelta del modo ottimale per elaborare i dati spesso è market- driven (interessi dell’industria) e/o gui data dal desiderio dei ricercatori di va lorizzare al massimo il proprio lavoro.
 e/o gui data dal desiderio dei ricercatori di va lorizzare al massimo il proprio lavoro..")
84
Possono esservi dubbi sulle modalità di applicazione dei risultati dei trial alla pratica clinica. Un problema frequente è : applicarli solo ai pazienti trial-like o anche ad altre categorie di pazienti ?

85
I problemi concernenti il trasferimen to rapido e appropriato dei risultati della ricerca alla pratica clinica hanno portato recentemente negli USA alla creazione di un certo numero di “Trans lational Medicine Institutes (TMIs)” de dicati a questo fine.
 de dicati a questo fine.")
86
Comitato etico esterno al trial IRB (Institutional Review Board)
")
87
L’IRB (Institutional Review Board, o Comitato Etico Locale) si occupa di tut- te le ricerche cliniche cui partecipa quel la data istituzione (Ospedale, Istituto universitario, etc.). Inoltre l’IRB si occu pa anche di altri aspetti bioetici per con to di quella istituzione : testamento bio logico, cure palliative, etc. Per quanto riguarda i trial clinici ran domizzati l’ IRB ne esamina il protocol lo, approvandolo o meno. A tal fine…

88
...oltre a vagliare i criteri generali di appropriatezza del protocollo (equipoi se, consenso informato, etc.), l’ IRB è chiamato a verificare che quel proto collo sia compatibile con la realtà loca- le : caratteristiche epidemiologiche, et niche, religiose degli arruolandi; strut ture operative dell’ istituzione; etc. L’IRB controlla poi lo svolgimento lo cale della ricerca e collabora con lo Ste ering Committee nei trial multicentrici.

89
Si noti che l’ IRB di solito non è in grado di analizzare approfonditamen te la letteratura medica pertinente (o altra eventuale documentazione) per ogni trial cui partecipa la propria isti tuzione ( soprattutto durante lo svolgi mento del trial) come può fare invece il DSMB, che si occupa solo di un trial…
 per ogni trial cui partecipa la propria isti tuzione ( soprattutto durante lo svolgi mento del trial) come può fare invece il DSMB, che si occupa solo di un trial…")
90
L’ Institutional Review Board può approvare o meno eventuali modifiche apportate in itinere al protocollo di un trial multicentrico (se non approva deve far ritirare la propria istituzione dallo studio), ma abitualmente non viene coinvol to nell’iter decisionale per l’eventu ale chiusura anticipata del trial.
, ma abitualmente non viene coinvol to nell’iter decisionale per l’eventu ale chiusura anticipata del trial.")
91
Concludo con due esempi di grave offesa alla correttezza del la ricerca clinica, l’ uno triste mente autentico, l’altro soltanto virtuale ma eloquente…

92
The code of silence Peter Wilmhurst, Lancet 1997; 349: 567-569 “Il Dott. John Darsee, cardiologo, lavorava nel dipartimento diretto da Eugene Braunwald ad Harvard. Egli aveva pubblicato un numero prodigioso di articoli scientifici, molti dei quali erano fraudolenti. Venne scoperto nel 1981…

93
Anche il Dott. Robert Slutsky, cardiologo in California, commi se analoghe frodi scientifiche, che furono scoperte nel 1985…

94
Nel novembre del 1982 Slutsky mi aveva detto di avere sempre pensato che i lavori di Darsee fos sero molto sospetti, dato che Dar see era il solo a pubblicare più di lui…”

95
HARLOT * How to Achieve positive Results with out Lying to Overcome the Truth * Prostituta

96
Articolo scherzoso (ma non troppo) di D. Sackett e A. Oxman, pubblicato nel BMJ del 20.12.2003, nel quale vengono offerti i servigi dell’ agenzia HARLOT agli sponsor industriali di trial clinici “ che non vogliano mettere a rischio l’accettazione dei loro prodotti affi dandosi all’ incertezza della scienza obiettiva”…

97
Nell’articolo vengono proposti tra l’al tro i Me-Too Protocols, con i quali, “ purchè il vostro farmaco anch’ io non sia molto peggio di un sorso d’acqua tridistillata, noi vi garantia mo un trial positivo” [Tanto sarcasmo non suscitò grandi ri mostranze. Evidentemente non manca vano buoni motivi per generarlo]

Presentazioni simili








Monitoraggio x verificare la fedeltà>")







>")


 di indagini diagnostiche, utilizzate.>")



>")